Cellular and Molecular Mediators of Neuroinflammation in Alzheimer Disease
Article information

Abstract
Alzheimer disease (AD) is a neurodegenerative disorder characterized by the loss of neuronal cells and the progressive decline of cognitive function. The major pathological culprit of AD is aggregation of amyloid-β (Aβ) and hyperphosphorylation of tau, eventually leading to progressive neuronal cell death and brain atrophy. However, the detailed molecular and cellular mechanisms underlying AD development as a result of neuronal cell death are little known. Although several hypotheses have been proposed regarding the development of AD, increasingly many studies suggest that the pathological progress of AD is not restricted to neuronal components such as Aβ and tau, but is also closely related to inflammatory responses in the brain. Abnormalities of Aβ and tau cause activity of pattern recognition receptors on the brain’s immune cells, including microglia and astrocytes, and trigger the innate immune system by releasing inflammatory mediators in the pathogenesis of AD. In this review, we present a basic overview of the current knowledge regarding inflammation and molecular mediators in the pathological progress of AD.
INTRODUCTION
Alzheimer disease (AD) is a neurodegenerative disease characterized by loss of neurons and synapses, leading to declines in learning and memory function in the brain [1,2]. The main hallmark of AD is the aggregation and deposition of amyloid-β (Aβ) peptides on the extracellular surface of neuronal cells, leading to the formation of Aβ oligomers and fibrils in the brain [3]. Another phenomenon observed in AD patients is hyperphosphorylation of tau protein in the brain, which accumulates in the microtubules of neurons and forms neurofibrillary tangles [4,5]. It is known that these 2 major hallmark features exert cytotoxic activities against neuronal cells, ultimately inducing the destruction of brain structure and memory decline [6,7].
Additionally, increasing evidence suggests that astrocytes and microglia are colocalized with Aβ plaques and neurofibrillary tangles in the brain of individuals with AD [8], implying that neuroinflammation may be a major component of AD pathogenesis. In epidemiological studies, AD patients who receive long-term treatment with an anti-inflammatory drug have shown diminished development of AD. Moreover, the correlation between genes regulating the immune response and AD pathogenesis has been confirmed by genome-wide association studies [9-11]. During the progression of AD, astrocytes and microglia are activated by the stimulation of Aβ plaques and neurofibrillary tangles. Activated astrocytes and microglia then migrate and surround the plaque and tangles, releasing inflammation-associated proteins such as cytokines, chemokines, and other pro-inflammatory mediators in the brain (Fig. 1) [12,13]. However, the cellular and molecular mechanisms underlying neuroinflammation in AD are not fully understood. Therefore, understanding the mechanisms that regulate neuroinflammatory processes and their impact on AD processes is important for the development of new strategies for AD treatment.
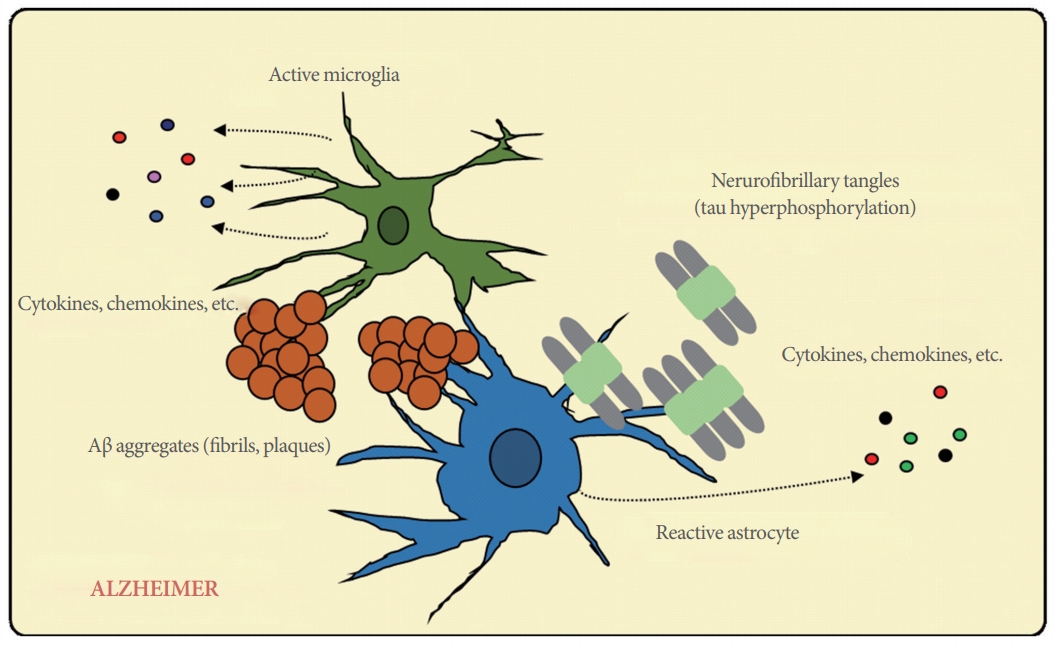
Neuroinflammation during Alzheimer disease development. In the presence of amyloid-β aggregates and neurofibrillary tangles, immune cells are activated and produce a variety of inflammatory mediators such as cytokines and chemokines.
CELLULAR MEDIATORS OF NEUROINFLAMMATION IN AD
Microglia
Microglia are the resident phagocytes of the brain, where they are ubiquitously present. They contribute to protection against infection by recognizing and responding to foreign antigens, while simultaneously supporting maintenance of the brain tissue [14]. To maintain these physiological functions, including microglial homeostasis, microglia express or release various molecules during brain development and in the normal adult central nervous system (CNS). In brain development, microglia regulate apoptosis of neuronal subpopulations by CD11b, triggering receptor expressed on myeloid cells 2 (TREM2), and DAP12; release neurotrophic factors such as brain-derived neurotrophic factor; and guide sprouting vessels. Microglia also contribute to maturation of the neuronal network and the maintenance of neuronal health by releasing CX3CR1 in the adult CNS [15].
Once activated by pathological triggers such as oxidative stress or misfolded protein aggregates, microglia begin to migrate to the locus of infection and initiate the innate immune response [16,17]. The initiation of the immune response by pathological triggers is mediated by receptor binding to pattern-associated molecular patterns or danger-associated molecular patterns. It has been suggested that the initial pathological trigger of microglial activation in AD is Aβ oligomers and fibrils, which are recognized by and bind with a variety of immune receptors including CD36, CD14, and toll-like receptors (TLR2, TLR4, TLR6, and TLR9) [18-21]. This binding of Aβ with immune receptors results in microglial activation, which induces the release of several pro-inflammatory cytokines and chemokines. It has been shown that removal of the immune receptor gene CD36 results in the reduction of Aβ-induced proinflammatory cytokine production and the prevention of intracellular Aβ deposition [22,23]. Aβ oligomers and fibrils are engulfed by the phagocytosis of activated microglia, and consequently undergo endosomal/lysosomal degradation processes for the clearance of Aβ [24].
In animal models of early AD development, the immune response induces Aβ clearance through the activation of microglia, indicating that the immune response favorably regulates AD-related pathologies [25-27]. However, chronic activation of the immune response by microglia results in an aggravation of AD pathologies, such as reactive microgliosis. The continuous activation results in sustained signaling transduction by pro-inflammatory cytokines, leading to neuronal damage and resulting in the loss of phagocytosis activity by microglia and diminished breakdown of Aβ plaques [28,29].
Further compelling evidence that compromised microglial function elevates the risk of AD through mis-regulation of the inflammatory response comes from studies identifying a rare mutation in the extracellular domain of TREM2 [30-32]. TREM2 is mainly expressed by the microglia and regulates the phagocytosis of Aβ. A rare mutation in TREM2 results in substantially increased AD risk [33-35] .
In the CNS of aging animals, microglial cells show an enhanced response to inflammatory triggers, similar to that observed in microglia in individuals with an ongoing neurodegenerative disorder [36,37]. Furthermore, microglia primarily have an immunomodulatory function and express many immune response-related antigens and molecules [38]. A recent study by Zare et al. [39] studied accumulation and effects of Aβ itself, suggesting these changes may reach beyond the CNS. A transgenic mouse model showed AD mice had immunoreactivity against Alzheimer’s disease markers in the bladder. These transgenic mice not only expressed Aβ in the bladder, but also these changes were associated inducing voiding dysfunction independent of the CNS, possibly through peripheral neurogenic means. However, the detailed mechanism of microglial function within the CNS remains debatable. Given that microglial activation continuously occurs, inducing innate and adaptive immune responses in the brain, further research will be needed to define the roles of microglia during AD pathogenesis.
Astrocytes
Astrocytes are the predominant glial cells observed in the CNS and play major roles in neuroprotection, organization, and maintenance in the brain. They are involved in multiple processes in the CNS, including neurotransmitter secretion and metabolism, synaptic remodeling, modulation of stress, neural information processing, and neuronal signaling transduction [40-42]. In early AD, similar to activated microglia, activated astrocytes are located around Aβ plaques and accompany the phagocytosis and degradation of Aβ, suggesting that they play an important role in the clearance of aggregated and accumulated Aβ in brain tissue affected by AD, along with microglia [13].
In AD animal models, the early response manifests by morphological changes including the atrophy of astrocytes, which may have functional consequences for synaptic connectivity. These changes have been shown to affect astrocytes located far from senile Aβ plaques in the later phase of AD progression [43-45]. Similar to microglia, astrocytes respond to fibrillar Aβ aggregates, which are responsible for the activation of astrocytes in brain tissue affected by AD. Reactive astrocytes then release many molecular mediators such as cytokines, nitric oxide, and other potentially toxic molecules, thereby enhancing the inflammatory response in the CNS. In an animal study, direct injection of Aβ oligomers strongly induced a significant activation of astrocytes via activation of the nuclear factor-kappa B (NF-κB) transcription factor and production of inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, S100, and cyclooxygenase-2 (COX-2). By activating astrocytes, NF-κB signaling tightly regulates the production of cytokines and chemokines, leading to neurodegeneration [46].
Oligodendrocytes
Oligodendrocytes are crucial for neurotransmission and the maintenance of neuronal morphology. It also has been established that oligodendrocytes are involved in immunological reactions in other neurological diseases, particularly multiple sclerosis. However, little is known regarding the functions of oligodendrocytes in the progression of AD [47,48]. A few studies have indicated that myelin abnormalities were found in the white matter of AD patients and that focal demyelination of axons was associated with Aβ aggregation in the gray matter of AD patients, as well as in the brains of AD transgenic mice [48,49]. Another study revealed that Aβ injections induced microglial proliferation, with attenuated damage to myelin and a functional loss of oligodendrocytes [50]. In an in vitro analysis, several types of Aβ peptides, such as Aβ (25-35), Aβ (1-40), and Aβ (1-42) induced cytotoxic effects on the oligodendrocytes [48,51,52]. It was also suggested that the differentiation and function of oligodendrocytes are affected by the PS1M146V mutation and Aβ deposition [53].
Finally, oligodendrocytes express mRNA of complement components such as C1q, C1s, C2, C3, C4, C5, C6, C8, and C9, leading to a complement-associated immune response in pathologically susceptible lesions of brain tissue affected by AD [54]. Therefore, complement-activated oligodendrocytes may an important target cell type in AD patients in whom inflammatory responses have been observed.
MOLECULAR MEDIATORS OF NEUROINFLAMMATION IN AD
Complement System
The complement system is an essential mechanism of the innate and adaptive immune response against pathogens. This system consists of cell surface proteins and proteases that are cleaved and activated in a cascade [55]. The complement system is divided into 3 pathways: (1) the classical pathway induced by the binding of antibody isotypes bound to antigens, (2) the alternative pathway induced by the binding of microbial cell surfaces in the absence of antibodies, and (3) the lectin pathway induced by the binding of mannose-binding protein, which binds to surface carbohydrates on microbes. During early steps of complement activation, complement components are sequentially cleaved by C3 convertase and C5 convertase in all 3 pathways. In the late steps of activation, C5b binds to C6, C7, C8, and C9 to form the membrane attack complex (Fig. 2) [56-59].
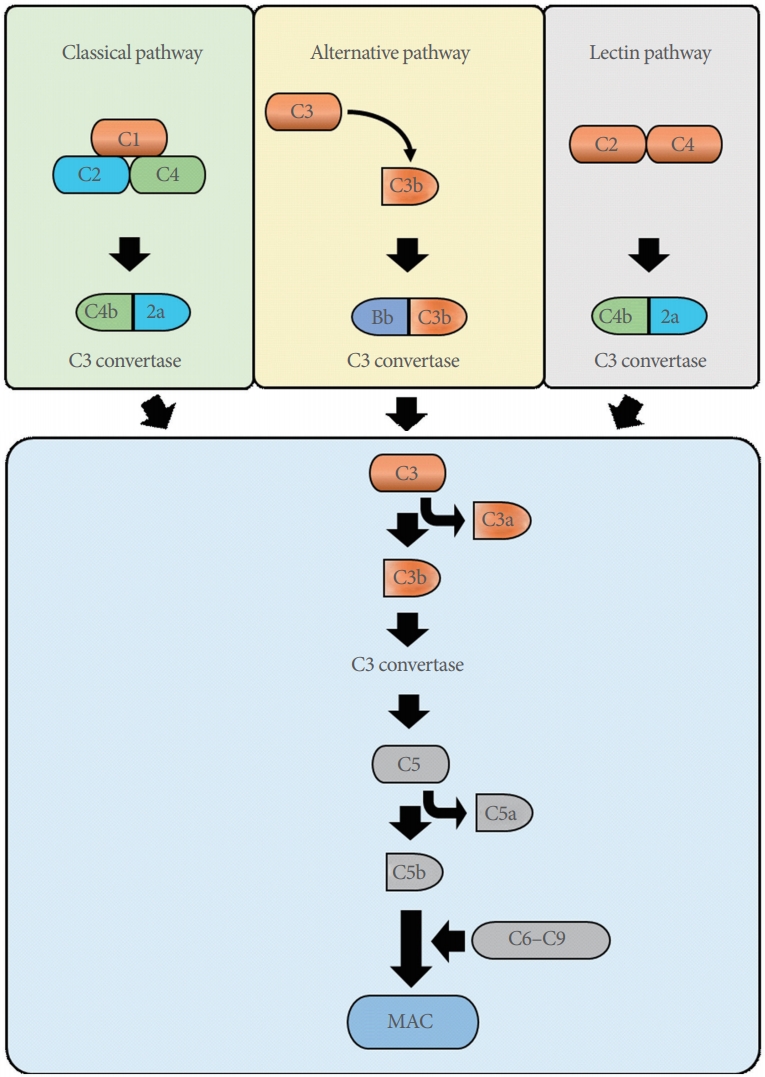
Schematic overview of the 3 complement pathways. MAC, membrane attack complex.
In studies of AD, amyloid precursor protein (APP) transgenic mice in which C3 activation is inhibited have shown increased Aβ accumulation. Consistent with this finding, increased Aβ deposition was observed in the brain of C3-deficient APP transgenic mice [25,60]. Moreover, activated complement components such as C1q have been reported to recognize the aggregated forms of Aβ, but not the monomeric forms, in vitro. An Aβ aggregate bound to C1q was found to be able to activate the alternative complement pathway, leading to processing and clearance of opsonized Aβ [61,62]. It appears that the activation of complement in AD might be effective for Aβ clearance; however, it also induces the production of neurotoxic materials by concomitant undesirable inflammation. Thus, additional studies will be required to provide convincing evidence of the function of the complement system in AD development.
Cytokines
Cytokines are mainly produced by microglia and astrocytes in the CNS and play a crucial role in the development of the CNS. Cytokines are involved in numerous inflammatory responses in neurodegenerative diseases [13]. Many studies of AD patients have revealed increased levels of pro-inflammatory cytokines, including TNF and IL [63,64]. In addition, several genetic investigations in mice showed that an elevated cytokine levels are significantly correlated with microglial activation and have effects on Aβ generation, neurodegeneration, and cognition [65,66].
First, TNF-α is one of the most important pro-inflammatory cytokines in AD, having beneficial or harmful functions on different neurons. High levels of TNF-α have been reported in the brains of AD patients [67]. Aβ directly stimulates TNF-α production from microglial cells through activation of the transcription factor NF-κB [68]. TNF-α also increases the expression of β- and γ-secretase, an enzyme involved in the generation of Aβ from APP in AD development [69,70]. In addition, mice lacking TNF receptor 1 crossed with the AD transgenic model showed reduced Aβ aggregation and microglial activation, along with a recovery of cognitive function [71].
Second, IL-1 is a major pro-inflammatory cytokine that is expressed in the early stage of Aβ deposition during AD development [72]. IL-1 is produced by microglial cells surrounding Aβ plaques and promotes the synthesis of S100, an inflammatory mediator, in astrocytes [73]. Within the IL-1 family, IL-1β production is strongly observed in the brain tissue of AD patients. IL-1β regulates the synthesis of APP, the secretion of APP from glial cells, and the amyloidogenic processing of APP [70]. Additionally, elevated levels of IL-1β in AD patients promote the activation of mitogen-activated protein kinase signaling, ultimately leading to the hyperphosphorylation of tau protein [74,75].
Finally, IL-6 is important for the normal homeostasis of brain tissue. Inhibition of IL-6 signaling promotes the reduction of microglial activation, while overexpression of IL-6 leads to chronic neuroinflammation [76]. In AD mouse models (TgCRND8 and Tg2576), overexpression of IL-6 in brain tissues has been observed [26]. Similar to IL-1β, IL-6 is also produced by microglial cells and results in elevated mRNA levels of the APP gene [77]. IL-6 has also been reported to induce the hyperphosphorylation of tau protein by increasing the CDK5 activator p53, resulting in the formation of neurofibrillary tangles, which play an important role in AD pathology [78].
Chemokines
Chemokines are a family of chemoattractant small cytokines that are mainly produced by astrocytes and microglia to regulate their migration to inflamed areas, enhancing neuroinflammation in AD development [79,80]. Significant changes in chemokines and their receptors are observed in the blood plasma, cerebrospinal fluid (CSF), and brain tissue of AD patients compared with otherwise healthy individuals [81,82]. It has been reported that most chemokines and their receptors contribute to the neuroinflammation involved in AD by engaging peripheral monocytes and promoting the activation of glial cells such as microglia and astrocytes [82].
Evidence of the cooperative role of chemokines in AD has been provided by the observation of upregulation of the chemokine receptors CCR3 and CCR5 in reactive microglia surrounding senile Aβ plaques [83,84]. A recent investigation of the CSF of AD patients revealed upregulation of CCL2, which was associated with cognitive decline [85]. Moreover, in vitro analyses have shown that Aβ promoted the generation of CXCR8, CCL2, CCL3, and CCL4 in astrocytes and microglia [86]. In AD mice, the neuronal death and cognition decline induced by Aβ deposition were found to be regulated by CX3CR1 and CX3CL1 [87-89]. Therefore, it has been suggested that in AD, chemokines are able to promote central and peripheral immunity, which contributes to disease progression.
Cyclooxygenases
Given that inflammatory mediators are closely associated with the pathology of AD [72], epidemiological studies have suggested that nonsteroidal anti-inflammatory drugs (NSAIDs), which are major inhibitors of COX, may be promising for AD drug development [90]. COX is an enzyme that is responsible for converting arachidonic acid in the process of prostaglandin synthesis. There are 2 types of COX: COX-1 and COX-2. Whereas COX-1 is expressed in many cell types and is involved in the physiological production of prostanoids, COX-2 is produced during inflammation and results in pro-inflammatory prostanoid synthesis [91,92]. COX-1 and COX2 are differently expressed in various stages of AD pathology [90]. While COX-1 is primary expressed in microglia, which are involved in Aβ aggregates in the late stage of AD, COX-2 is highly expressed in neurons and is colocalized with the expression of cell cycle proteins under conditions of low Aβ deposits and tau tangles in the early stage of AD.
In AD mice models, overexpression of COX-2 in neurons contributed to neuronal cell death by the formation of Aβ plaques and by the production of free radicals, causing aggravated cognitive deficits. Furthermore, the appearance of neuronal death in AD was inhibited by treatment with NSAIDs [91-95]. Thus, many scientists have suggested that drug development using NSAIDs to target COX-mediated neuronal cell death may be a promising potential strategy for the treatment of AD.
CONCLUSIONS
Here, we investigated neuroinflammation and consequent inflammatory mediators (cellular and molecular) in the pathological progress of AD. Inflammation is not only found in many tissues and lymphoid organs, but is also observed in neurodegenerative diseases such as AD. Many scientists have proposed that inflammation occurs in the presence of misfolded Aβ and tau proteins, resulting in initiation or acceleration of the development of the disease. However, other investigators have argued that inflammation might be a beneficial defense mechanism against neurotoxicity in brain tissue affected by AD. Whether inflammation promotes or alleviates AD, it should be acknowledged that neuroinflammation plays a major role in the development of AD. Therefore, additional studies should be conducted to define the detailed molecular mechanisms and crosstalk between neuroinflammation and AD. Therapeutic approaches targeting and regulating neuroinflammation will be a promising frontier in terms of new treatments for AD.
Notes
Fund/Grant Support
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2015R1A6A3A04058568).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.