NGS TECHNOLOGIES AND PLATFORMS
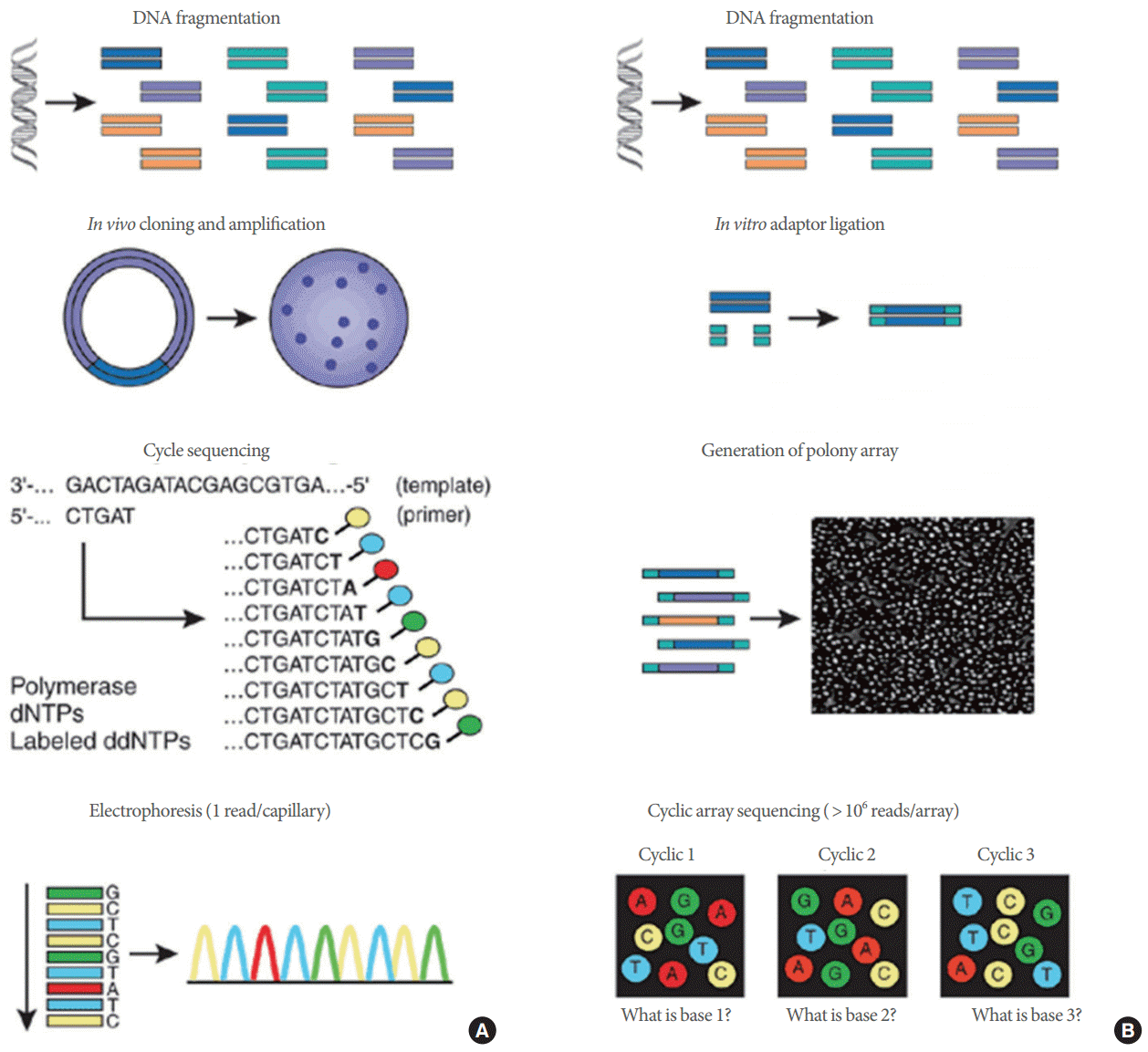
Next-generation sequencing (NGS) technologies are methods that sequence nucleotides faster and cheaper than Sanger sequencing. These massively parallel DNA sequencing methods have opened a new era of genomics and molecular biology. Compared to the traditional Sanger capillary electrophoresis sequencing method [1,2], which is considered a first-generation sequencing technology, NGS technologies provide higher throughput data with lower cost and enable population-scale genome research. NGS technologies have three major improvements compared to first-generation sequencing [3]. First, NGS methods do not require a bacterial cloning procedure and prepare libraries for sequencing in a cell free system. Second, NGS technologies process millions of sequencing reactions in parallel and at the same time. Third, detection of bases is performed cyclically and in parallel.
These major improvements allow scientists to process sequencing of entire genomes with low cost and in a very short period of time. Fig. 1 shows the overall workflows of conventional sequencing and NGS [4]. However, NGS technologies needed the development of novel alignment algorithms to assemble and map the genome from the relatively short reads [5]. As of 2016, there are 3 major platforms of NGS technologies. Roche (formerly 454) has the most recent 454-based sequencer, GS FLX+ (Roche Diagnostics Co., Branford, CT, USA), which generates about 1,000,000 reads of 700 base pairs (bp) [6]. Life Technologies, part of Thermo Fisher (Waltham, MA, USA), has the Personal Genome Machine and Proton with their Ion torrent technology. Proton uses semiconductor-sequencing technology with solid-state pH meters detecting a hydrogen ion released from DNA on chip. Proton generates 60–80 million reads of up to 200-bp fragments, which delivers up to 10 Gb of sequence per run with an Ion P1 Chip [7]. Illumina (San Diego, CA, USA) acquired Solexa in 2007 and is supplying most NGS platforms in the world such as HiSeq2500, HiSeq4000, and HiSeq X. Illumina’s the most recently available high-throughput sequencer is the HiSeq X TEN system consisting of 10 HiSeq X. The HiSeq X delivers 1.8 Tb of sequence per run in 3 days from ~6 billion reads of 150 bp and is especially designed for whole genome sequencing (WGS) that requires ultrahigh throughput and multiparallel sequencing at the same time [8]. This system may be able to break US$1,000 per human WGS. This is almost a 10,000-fold reduction in the cost of sequencing a human genome since 2004 (Fig. 2) [9].
Other NGS technologies have been developed by companies like Qiagen, Helicos Biosciences, and Pacific Biosciences (now part of Roche). These platforms sequence directly from template DNA, while the previously discussed NGS technologies amplify the DNA template during the library preparation steps. Thus, these technologies are called third-generation sequencing technologies (to distinguish from NGS technologies). The leader of the third-generation sequencing field is Roche. Roche has released PacBio RS II from Pacific Biosciences technology (Menlo Park, CA, USA), and the PacBio RS II generates several thousands of long reads with up to 20,000 bp [10]. This long-reads sequencing technology has an advantage in de novo genome assemblies because the contig and scaffold N50 values are substantially higher than de novo genome assemblies by short-reads sequencing [11,12].
APPLICATIONS OF NGS
There are many applications for NGS and new methods being developed continuously. There are several classifications for NGS applications. In this section, we classify NGS applications according to the experimental purpose.
(1) To build a new genome from unknown organisms, researchers use de novo sequencing with assembly. This de novo genome assembly requires a tool called an “assembler.” Assemblers put fragmented reads of DNA together like a jigsaw puzzle by aligning regions with overlap to build a genome sequence [13].
(2) To measure genetic variation from an organism with an existing reference genome, researchers can do DNA-sequencing, RNA-sequencing, and epigenome sequencing. In the case of DNA-sequencing, whole genome, whole exome (for eukaryotes), and targeted sequencing are available with NGS technologies. By comparing sequencing results to reference genomes, researchers can see the genetic variation such as single nucleotide polymorphisms (SNPs), structural variations, copy number variations, and other variations using various software programs [14].
(3) To analyze transcriptome results with sequencing, researchers synthesize complementary DNA from RNA for sequencing (There are RNA preparation library kits on the market for NGS platforms.) RNA sequencing allows researchers to examine splicing of RNA, gene fusion, mutation, and differential gene expression. Compared to the hybridization-based microarrays for gene expression studies, microarrays show artifacts of hybridizations, a narrow range of expression quantitation, low resolution from several to 100 bp, and limitation of coverage based on probes [15]. The technical advantages of RNA-Seq has led to a transition in transcriptomics from microarrays to sequencing-based methods.
(4) For epigenome studies and regulatory mechanisms of the genome, researchers can use DNA methylation sequencing and chromatin immunoprecipitation followed by sequencing (ChIP-Seq). To determine methylation of CpG dinucleotides, the bisulfite sequencing method is applied. Bisulfite treatment converts cytosine residue to uracil so only the methylated cytosine residues are detected. ChIP-Seq is a method for analyzing protein-DNA interactions, such as the binding sites of transcription factors. ChIP-Seq requires antibodies for proteins of interest to enrich the DNA regions bound by proteins in living cells. Several research publications have used ChIP-Seq to demonstrate and predict genome-wide networks of regulation [16,17].
(5) NGS technologies allow microbial ecology scientists to investigate genetic materials from environmental samples on a tremendous scale. Scientists can use extracted DNA from environmental samples without cloning [18].
As described above, NGS technologies provide opportunities for better quality and quantity to scientists in many fields. Many other new methods with NGS and preparation technologies are being introduced now.
HUMAN GENETICS AND GENOMICS
Human genetics is the study of inheritance in human beings and encompasses various fields including classical genetics, cytogenetics, genomics, population genetics, and clinical genetics. To study human genetics for many purposes, researchers wanted to create a fully mapped sequence of the human genome and initiated the human genome project (HGP) in 1990. The HGP aimed to map the nucleotides in a human haploid reference genome. The HGP was completed in April 2003 and finally published on May 27, 2004 [19]. The sequence is now stored in databases available to anyone on the internet. The National Center for Biotechnology Information (NCBI) built a database known as GenBank [20] and other organizations including the University of California, Santa Cruz and Ensembl [21] present additional data with powerful tools for search and visualization in the UCSC Genome browser [22] and Ensembl Browser [21], respectively. The human reference genomes are maintained by the Genome Reference Consortium (GRC), which is an international collective of academic and research institutes from the HGP. The current reference genome is GRCh38.p8, which was released on June 30, 2016. This is an updated release from GRCh38, released on December 24, 2013 (new human genome assembly [GRCh38] released) [23].
The introduction and low cost of new technologies in sequencing is leading to an era of personal genome sequences. Many of the new human genome sequencing projects from individuals have reported additional human genome sequences. Comparison studies with the original human reference genome have shown diversity in human genetic variation by ethnicity and regions of ancestry [24-28]. Some examples follow.
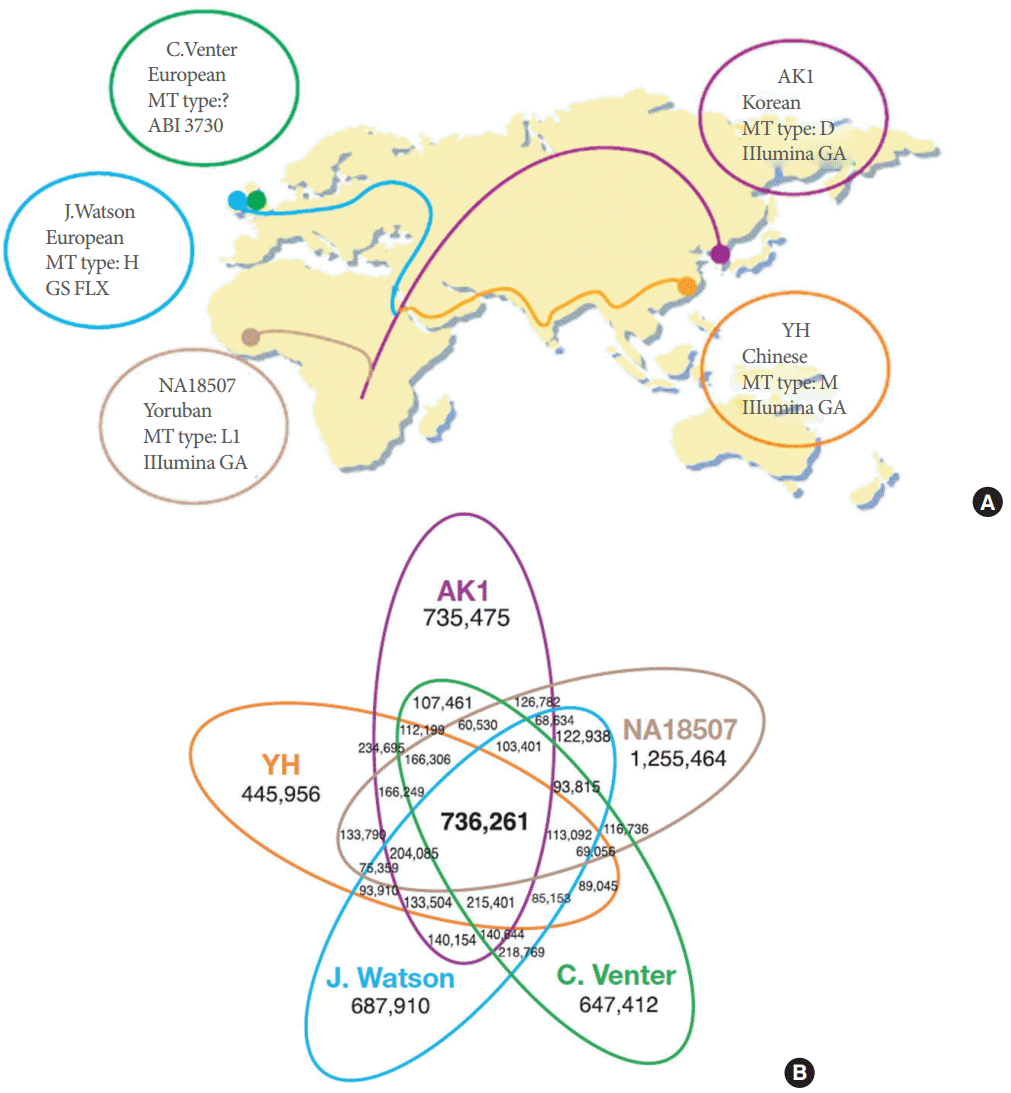
(1) One of the comparison studies showed an indel distribution pattern among 5 different sequenced genomes [28]. Five sequenced genomes from 3 different distinct geographic regions including the newly sequenced AK1 from a Korean individual (Fig. 3A). The possibility of differences in technical procedures or interindividual variability was explained. Bioinformatic analysis for SNP detection from aligned sequences showed that 21% of AK1’s SNPs were unique and 8% were identical in all genome sequences (Fig. 3B). Many other individual genome sequences are being reported continuously, and one report noted that South Asians have a higher risk of type-2 diabetes and cardiovascular disease compared to Europeans, which is the current human reference genome [29]. These reports explained that the genome of each individual is unique and emphasized the necessity of personal genome sequencing and population genomics to identify genome sequences possibly related with diseases in medical genetics.
(2) Population genomics is the large-scale comparison of DNA sequences of populations. This is a neologism and a new paradigm in population genetics by combining genomics concepts and technologies [30]. Population genomics uses genomewide sampling to identify the phenotypic variation such as gene flow and inbreeding and to improve understanding on microevolution [31,32]. In human population genomics, there was a revolution with recent advancements in sequencing and data analysis. These advancements allowed scientists to study hundreds and thousands of loci from populations and enable genome wide effects and/or focus with genome-scale data.
(3) Medical genetics is the branch of medicine involving the diagnosis and management of hereditary disorders. Medical genetics considers the diagnosis, management, and counseling people with genetic disorder as a form of medical care, while research-oriented human genetics focuses on the causes and inheritance of genetic disorders. Clinical genetics and cancer genetics are the practices of clinical medicine for hereditary disorders and cancers, respectively. Also, clinical and cancer genomics are new fields with genome sequencing to inform patient diagnosis and care by diagnosing genetic diseases, categorizing patients for appropriate treatment, and providing information about an individual’s response to treatment. However, clinical and cancer genomics require a more comprehensive view of genomics information and associated biological implications [32]. It is believed that the convergence of research-based genomic research and clinical/cancer genomics for medical care will become increasingly important.
NEW ERA OF WGS
WGS follows the current trends in the convergence of fundamental genomic research and clinical implications of the presence or absence of certain genes. Now, WGS is becoming one of the most widely used applications and is providing tremendous quantities of genome sequences relative to the past through public and private human genome sequencing projects throughout the world. As of 2015, the genomes of 2,504 people from 26 different populations have been reconstructed according to reports [33,34] from the 1,000 Genomes Project [35].
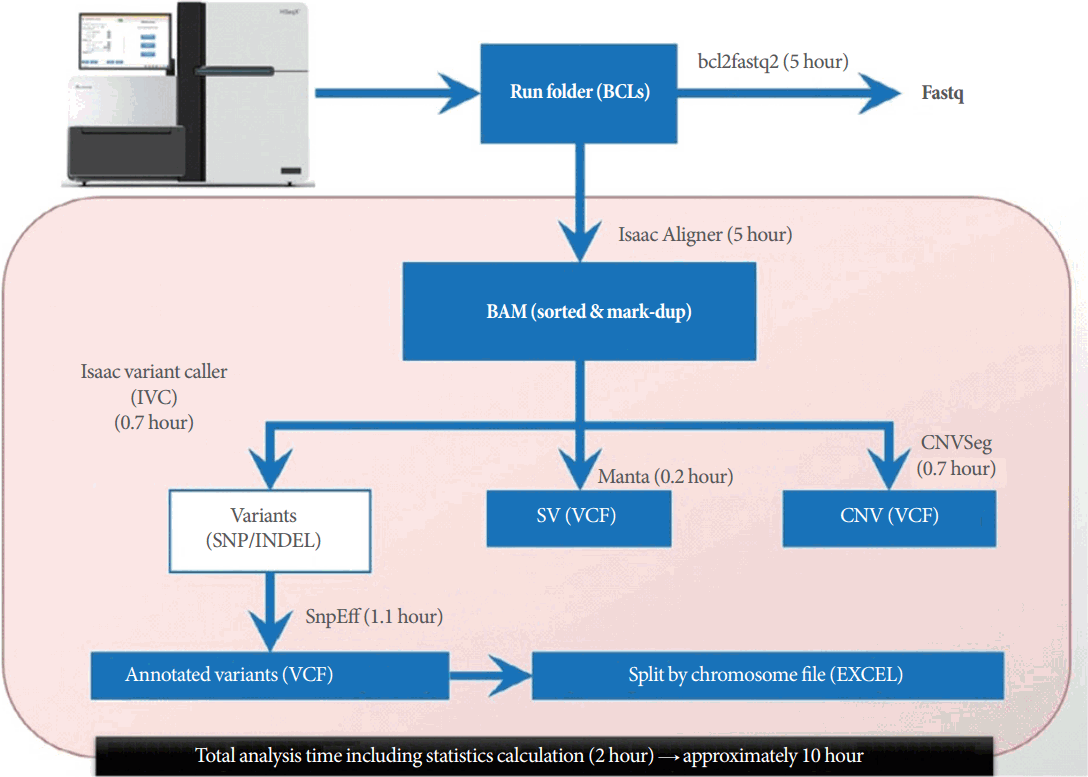
The release of HiSeq X Ten systems with a capacity of 1.8-Tb sequences per run brought a new era of WGS explosively by the reduction of costs. These HiSeq X Ten systems can deliver 18,000 Genomes a year. The National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (NIH) planned and processed 20,000 Genomes for their TOPMed (Trans-Omics for Precision Medicine) program by 2015 and has expanded the WGS project with 62,000 individual genomes. Besides NHLBI/NIH, many nonprofit and profit organizations are initiating and expanding their WGS project to build and understand individuals’ genomes. Researchers are expecting to discover molecular biomarkers, identify potential drug targets, enable clinical trials, and accelerate systems medicine and emerging precision medicine for predicting, preventing, diagnosing, and treating diseases [36]. Fig. 4 shows a WGS workflow using the HiSeq X and analysis pipeline.
There are several interesting human genome sequencing projects that continue this research throughout the world.
(1) The Human Genome Project–Write (HGP-Write) is a ten year extension of the HGP to synthesize the human genome (Science June 2, 2016 and The Scientist, June 2, 2016). HGP revealed that the human genome consists of 3 billion DNA nucleotides and HGP-Write will try to synthesize large portions of the human genome for scientific and medical advances [37,38]. This project will be managed by the Center of Excellence for Engineering Biology, a new nonprofit organization.
(2) The 100,000 Genomes Project is continuing the sequencing project from the 1,000 Genome Project. Genomics England, a government-owned company, has introduced this new 100,000 Genome Project to sequence 100,000 whole genomes from National Health Service (NHS) patients and their families. The aim of this project is to create a new genomic medicine service for the NHS and to enable new medical research that combines genomic sequence data with medical records. Researchers will study the best way to use and interpret genomic data for healthcare and investigate the cause, diagnosis, and treatment of diseases [39].
(3) The most recent human genome project is GenomeAsia 100K (GA100K). GA100K is a mission-driven nonprofit consortium consisting of Nanyang Technological University (NTU), Macrogen, and MedGenome. NTU is a research-intensive public university and has a new medical school, the Lee Kong Chian School of Medicine, set up jointly with Imperial College London. Macrogen is a world leading genetic service provider with global locations including Korea, the United States of America, Japan, and The Netherlands, and a spinoff company from the Genomic Medicine Institute in Seoul National University. MedGenome is a genomic-driven research and diagnostic company and the market leader of genetic diagnostic testing in India. This consortium will collaborate to sequence and analyze 100,000 Asian individuals’ genomes to help accelerate population specific medical advances and precision medicine. Asians are significantly underrepresented in current genomic studies with Caucasian based human genome reference and databases even though there are unique genetic differences between South and East Asians. GA100K plans to create reference genomes for Asian populations as well as identify population-specific alleles. With this project, GA100K expects to understand biology of diseases and identify new possible therapeutic drugs [40].
CLOSING REMARKS AND PROSPECTIVE
In this short review article, we summarized and discussed the current trends of genomics and the reference genomes being built with new NGS technologies that can be applied to personalized and precision medicine. NGS technologies have enabled scientists to interrogate biological systems with population-scale genomics and have been increasingly popular for biological and clinical research. The low cost and high throughput data of NGS technologies provide more opportunities in human genetics and genomics along with newly developed data algorithms. Sanger sequencing initiated HGP, which provided a human reference genome with huge amounts of data. The 1,000 Genome Project generated genome data from 2,504 individuals in 26 different populations for 5 years. The most recent NGS instrument, the HiSeq X Ten, allows researchers to have 20,000 genomes available from the NHLBI TOPMed program in 2015. This trend in human genome research initiated the Precision Medicine Initiative by the U.S. government [41] and other, similar projects/programs by countries and profit/nonprofit organizations have also been progressing. The accumulation of human genome data and have demonstrated the importance of appropriate human reference genomes as an aspect of personalized medicine. The 100,000 Genome Project and GenomeAsia 100K are major projects for accelerating specific population-scale medical advances and precision medicine.
Third-generation sequencing technologies have been introduced and used mostly at the research level. There are some studies that compare NGS and third-generation sequencing technologies [42]. So far, research is still more dependent on NGS technologies, and third-generation sequencing technologies are aiding and/or supplementing NGS methods. However, we may expect another paradigm shift in genomics in the near future.