The Role of NLR-related Protein 3 Inflammasome in Host Defense and Inflammatory Diseases
Article information
Abstract
Among a number of innate receptors, the nucleotide-binding domain leucine-rich repeat containing (NLR) nucleotide oligomerization domain (NOD)-like receptor families are involved in the recognition of cytosolic pathogen- or danger-associated molecules. Activation of these specific sets of receptors leads to the assembly of a multiprotein complex, the inflammasome, leading to the activation of caspase-1 and maturation of the cytokines interleukin (IL)-1β, IL-18, and IL-33. Among NLRs, NLR-related protein 3 (NLRP3) is one of the best-characterized receptors that activates the inflammasome. There is no doubt that NLRP3 inflammasome activation is important for host defense and effective pathogen clearance against fungal, bacterial, and viral infection. In addition, mounting evidence indicates that the NLRP3 inflammasome plays a role in a variety of inflammatory diseases, including gout, atherosclerosis, and type II diabetes, as well as under conditions of cellular stress or injury. Here, we review recent advances in our understanding of the role of the NLRP3 inflammasome in host defense and various inflammatory diseases.
INTRODUCTION
The mammalian immunity has effective systems for fighting against pathogenic invasion, namely, the innate and adaptive immune systems. The innate immune system comprises molecules, cells, and mechanisms that play important roles in fighting bacterial and viral infection and act as the frontline of immune defense. The activation of innate immunity can lead to the induction of the adaptive immune system through a process of antigen presentation [1]. Upon challenge by pathogens or other dangers, the innate immune system recognizes warning signals by sensing pathogen- or danger-associated molecular patterns (PAMPs or DAMPs, respectively) by way of innate receptors. Although it was thought for a long time that the innate immune system is non-specific to particular antigens, it is now understood that it can discriminate self from non-self through pattern recognition receptors (PRRs), which include membrane-bound and cytosolic receptors [1,2]. The last several decades of study have enriched our knowledge of the innate receptors and how these receptors recognize and respond to pathogenic and modified self (i.e., danger) signals.
The nucleotide-binding domain leucine-rich repeat (LRR) containing (NLR) family of cytosolic receptors plays an important role in the formation of multiprotein complexes, i.e., inflammasomes, which are able to activate the cysteine protease caspase-1 and induce the maturation of interleukin (IL)-1β and IL-18. IL-1β is a cytokine that plays a crucial role in host defense and inflammation. The secretion of IL-1β is very tightly regulated by an inflammasome that comprises NLR, an intracellular receptor, and apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain (CARD) (ASC). The activation of this inflammasome can lead to biological activation of IL-1β from its inactive proform to active IL-1β by catalysis of the enzyme caspase-1, which is itself activated by the inflammasome [3]. The NLRP3 inflammasome is the most intensively studied of the inflammasomes, and it can be activated by a variety of microbial-, stress-, or danger-induced substances [4]. However, the ligands and exact activating mechanism(s) of the NLRP3 inflammasome remain to be identified [4].
Compelling evidence has shown that the inflammasome is important for pathogenesis and/or regulation of a variety of inflammatory diseases. Currently, therapeutic approaches involving anti-IL-1β strategies are known to be successful for the treatment of auto-inflammatory (IL-1β activation disorders) and inflammatory diseases [5]. A better understanding of the specific nature of the NLRP3 inflammasome is important to reveal the molecular pathophysiology of various inflammatory diseases and to identify potential therapeutic modalities to control inflammasome activation. In this review, we summarize recent reports of the roles of the NLRP3 inflammasome in host defense and inflammatory disorders, including gout, atherosclerosis, and type II diabetes. Finally, we discuss the regulation of IL-1β secretion and NLRP3 inflammasome activation and highlight their therapeutic potential.
OVERVIEW OF NLRP3 INFLAMMASOME ACTIVATION
NLRP3 (Nucleotide binding oligomerization domain [NOD/NACHT], LRR, and PYD domains [PYDs] containing protein 3; also known as NALP3, cryopyrin, or CIAS1), a member of the NLR family, interacts with the adaptor molecule ASC (also called PYD and CARD domain containing PYCARD) and caspase-1 to form a large (>700 kDa) multiprotein complex called the NLRP3 inflammasome [3,4,6]. Activation via PAMPs or DAMPs enables the N-terminus of NLRP3, which contains a PYD, to mediate homotypic interactions with ASC via PYD-PYD interactions, which in turn permits the subsequent recruitment of pro-caspase-1 via an interaction between CARD-CARD and ASC [7], leading to the autocatalytic activation of caspase-1 (Fig. 1).
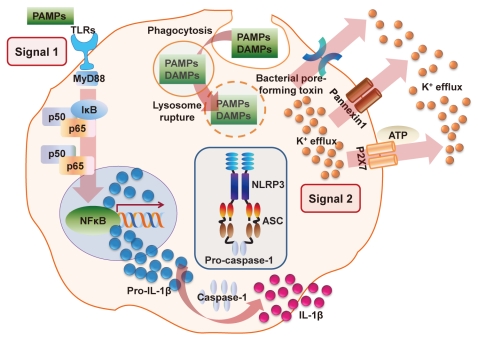
A schematic model for NLR-related protein 3 (NLRP3) inflammasome activation. The activation of NLRP3 leading to the secretion of interleukin (IL)-1β and IL-18 is triggered by a number of stimuli. Diverse pathogen-associated molecular patterns (PAMP) and/or danger-associated molecular patterns (DAMP) stimulation potentiates two steps that activate the NLRP3 inflammasome. Signal 1 activation leads to the expression of the pro-IL-1β gene and production of the pro-IL-1β protein through the toll-like receptor (TLR)-MyD88-NFκB signaling pathway. Signal 2 is a critical step in inflammasome activation. These signals or agonists trigger the assembly of a large macromolecular complex through the recruitment of the apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain (CARD) (ASC) adaptor protein and pro-caspase-1 to NLRP3. Several mechanisms have been suggested for NLRP3 inflammasome activation, including pore formation through P2X7 receptor and K+ efflux, mitochondrial reactive oxygen species generation, phagocytic pathway activation by particulate or crystalline structures (e.g., monosodium urate crystals, aluminium potassium sulfate, or silica nanoparticles), and lysosome rupture, among others. The molecular mechanisms by which NLRP3 inflammasome activation occurs are not yet fully understood. ATP, adenosine triphosphate.
The NLRP3 inflammasome activates caspase-1 by a two-step process. An initial priming step (signal 1) is required for subsequent NLRP3 inflammasome formation by an activating signal (signal 2) [8,9]. In the absence of NLR signals, PRRs (such as toll-like receptor 4/lipopolysaccharide [TLR4/LPS]), cytokine receptors, or other signaling factors (signal 1) lead to the upregulation of pro-IL-1β or pro-IL-18 transcription. In response to a variety of intracellular signals (signal 2), such as endogenous danger signals (for example, purinergic P2X7 receptor [P2X7R]/adenosine triphosphate [ATP]) and bacterial ligands, NLRs are released from their auto-inhibited monomeric conformation, leading to the assembly of an inflammasome capable of activating caspase-1 [9-12]. Once activated, caspase-1 processes the precursor forms of IL-1β and IL-18 to generate the biologically active forms of these proinflammatory cytokines that are then released from the cell [7,8].
The NLRP3 inflammasome assembles in response to a variety of exogenous and endogenous activators that include microbial signals from bacteria, fungi, or viruses that contain single-stranded (ss) RNA or double-stranded (ds) RNA analogs, bacterial pore-forming toxins (e.g., nigericin), environmental irritants (e.g., asbestos, silica, alum, or ultraviolet light), endogenous danger signals (e.g., ATP, monosodium urate [MSU] crystals, β-amyloid, or hyaluronan), crystalline substances, and peptide aggregates [8,12-15], as well as extracellular ATP released from dying tumor cells [16]. However, the molecular interactions that engage the relatively non-specific NLRP3 inflammasome in response to such distinct stimuli are unclear at present.
There are a number of potential mechanisms for the assembly of the NLRP3 inflammasome. According to one hypothesis, mitochondria are the principal source of reactive oxygen species (ROS) required for inflammasome activation; several recent studies have implicated ROS produced by mitochondria, rather than phagosomes, in NLRP3 activation [17,18]. A second mechanism involves the disruption of lysosomal membrane integrity by crystalline materials and peptide aggregates [19,20]. Upon uptake of such substances, lysosomal rupture leads to the leakage of lysosomal proteases, specifically cathepsins B and L, into the cytosol where they could possibly mediate NLRP3 inflammasome activation by an as-yet-undefined cleavage event. It is probable that both mechanisms are required for full activation of the NLRP3 inflammasome or that they operate co-dependently. Cellular potassium efflux has also been shown to be a requirement for inflammasome activation [21]. Given the importance of NLRP3 inflammasome activation for inflammation, complex activation mechanisms and elaborate regulatory processes are expected.
THE NLRP3 INFLAMMASOME AND HOST DEFENSE
The products of NLRP3 inflammasome activation, IL-1β and IL-18, have been shown to be protective in a variety of infections [22,23]. We first discuss the involvement of the NLRP3 inflammasome in host defense against fungal, bacterial, and viral infections.
Fungal Infection and the NLRP3 Inflammasome
In immunocompromised patients, some fungal infections, as opportunistic pathogens, are often fatal. It has not been shown that the NLRP3 inflammasome is involved in susceptibility to fungal infections in humans; however, studies using murine models have established its role in host defense and resistance to several fungal infections, including Candida albicans, Aspergillus fumigatus, and Saccharomyces cerevisiae [15,24-27]. Studies have also shown that the inflammasome adaptor protein, ASC, and sensor protein, NLRP3, are important for caspase-1 activation and IL-1β secretion in response to the conserved fungal components zymosan, mannan [7], and large particulate (1,3)-β-glucans [28]. Interestingly, the yeast-phase forms of A. fumigatus and C. albicans induce lower activation of the NLRP3 inflammasome, reflecting the differential regulation of host defense responses that depend on the morphological form of fungi [24,25,27]. The Dectin-dependent Syk kinase signaling pathways are required for upregulation of pro-IL-1β at the transcriptional level and inflammasome activation by C. albcans [15] or A. fumigatus [27], suggesting that Syk kinase signaling mediates NLRP3 inflammasome activation.
Bacterial Infection and the NLRP3 Inflammasome
The role of the NLRP3 inflammasome in infections with bacterial pathogens has been widely studied. In infections with Gram-positive strains, such as Staphylococcus aureus or Listeria monocytogenes, IL-1β secretion is induced in macrophages through NLRP3 inflammasome activation [9,29]. In addition, Streptococcus pyogenes activates the NLRP3 inflammasome through NF-κB and the virulence factor streptolysin O, but this activation does not require exogenous ATP or the P2X7R protein [30].
The Gram-negative pathogens Vibrio vulnificus and V. cholera have been reported to trigger the activation of caspase-1 and IL-1β secretion in macrophages via NLRP3 inflammasome activation [31]. Salmonella typhimurium, a Gram-negative flagellated bacterium, induces the activation of the NLRC4/IPAF inflammasome [14,32]. S. typhimurium can also induce NLRP3 inflammasome activation via Salmonella pathogenicity island (SPI)-2-dependent mechanisms. Moreover, both NLRs, NLRP3 and NLRC4, are necessary for recruitment of ASC and caspase-1 and activation of pro-IL-1β processing. Consistently, mice harboring both Nlrc4 and Nlrp3 genetic defects are more susceptible to Salmonella infection [33]. Recent studies have documented the protective roles of NLRP3 and NLRC4 in infections with Burkholderia pseudomallei, a gram-negative bacterium that causes melioidosis [34]. Although mice deficient in NLRP3 and NLRC4 are susceptible to infection with B. pseudomallei, the NLRP3 and NLRC4 inflammasomes have non-redundant roles in melioidosis [34]. NLRC4 is essential for pyroptosis, while NLRP3 regulates the production of protective IL-18 in infections with B. pseudomallei [34].
Neisseria gonorrhoeae, the pathogen that causes the sexually transmitted disease gonorrhea, can induce IL-1β production via NLRP3 inflammasome activation and pyronecrosis, which are dependent on the virulence factor lipooligosaccharide [35]. In this case, the activation of NLRP3-induced inflammatory responses and host cell death contribute to the pathogenesis of N. gonorrhoeae [35]. During infection with another sexually transmitted infectious pathogen, Chlamydia trachomatis, IL-1 may be involved in the development of oviduct pathology, because the presence of an IL-1β antagonist prevents C. trachomatis-mediated cytotoxicity and tissue destruction in a fallopian tube organ culture model [36].
It is evident that inflammasome activation is differentially modulated in specific cell types and/or different species. For example, Francisella tularensis infection leads to absent in melanoma 2 (AIM2) inflammasome activation in an exclusive manner [37]. However, recent studies have shown that Francisella can activate the NLRP3 inflammasome in human cells through ROS, cathepsin B, and potassium efflux pathways [38]. In murine macrophages, Mycobacterium tuberculosis prevents inflammasome activation and IL-1β maturation through the mycobacterial gene zmp1, which encodes a putative Zn(2+) metalloprotease [39]. However, in human macrophages, the M. tuberculosis ESAT-6 protein can potently activate the NLRP3/ASC inflammasome [40]. Recent studies have shown that the NLRP3 inflammasome activated by M. tuberculosis does not directly promote host defense responses [41] or susceptibility to active tuberculosis [42], but is involved in necrotic cell death during mycobacterial infection [43]. Among numerous atypical mycobacteria, M. abscessus can activate the NLRP3 inflammasome through Dectin-1-Syk-dependent signaling pathways [44]. The activation of the NLRP3 inflammasome plays an essential role in antimicrobial responses against M. abscessus in human macrophages [44]. However, in pathogenic M. marinum infection, the Esx-1 (type VII) secretion system promotes the activation of the NLRP3 inflammasome, which exacerbates disease and plays a host-detrimental role during infection [45]. A better understanding of the functions of the NLRP3 inflammasome during bacterial infection is required to clarify the contribution of this essential protein complex to host-pathogen responses, particularly in the context of innate and pathophysiologic responses during infection.
Viral Infection and the NLRP3 Inflammasome
The NLRP3 inflammasome is required for recognition of several RNA viruses, including influenza and encephalomyocarditis viruses (EMCV), whereas the retinoic acid-inducible gene I (RIG-I) inflammasome plays a role in detection of vesicular stomatitis virus (VSV) [8,46,47]. Besides viral dsRNA and its analog poly (I:C), Sendai virus and influenza virus infections are known to activate the NLRP3 inflammasome and the production of active IL-1β and IL-18 in macrophages [8]. Recently, it was found that IL-1β production during respiratory syncytial virus infection is caspase-1-dependent, and that NLRP3 inflammasome activation is required for IL-1β release in virus-infected cells [48]. Another recent report showed that EMCV and VSV can induce NLRP3 inflammasome activation through melanoma-differentiation-associated gene 5- or retinolic acid inducible protein I-independent mechanisms [46]. In that study, there was no significant difference in susceptibility to viral infections between wild-type and caspase-1-deficient mice, suggesting that the NLRP3 inflammasome is not critically involved in host defense against these viral pathogens [46]. In human hepatoma cells, hepatitis C virus infection induces the assembly of the NLRP3 inflammasome complex and activation of IL-1β through ROS-dependent mechanisms [49].
The AIM2 protein is an important sensor for cytosolic DNA that is in a complex with ASC [50]. However, certain DNA viruses have been shown to activate the NLRP3 inflammasome in vitro and in vivo. Varicella-zoster virus (VZV), an alphaherpes DNA virus that is the causative agent of varicella and herpes zoster, can induce formation of the NLRP3 inflammasome [51]. The VZV-induced activation of the NLRP3 inflammasome does not require ROS or AIM2 expression [51]. Modified vaccinia virus Ankara, an attenuated dsDNA poxvirus, activates NLRP3 inflammasome pathways for expression and processing of IL-1β [52]. Although these data indicate that the NLRP3 inflammasome is activated upon detection of a variety of RNA viruses and certain DNA viruses, its precise roles in host defense or pathology remain to be determined. Interestingly, recent reports have shown that 3'-untranslated region NLRP3 polymorphisms are linked to susceptibility to human immunodeficiency virus-1 infection, although functional analysis has not been performed [53]. It is necessary to determine whether certain viral infections are associated with NLRP3 or ASC genetic polymorphisms to clarify the role(s) of the NLRP3 inflammasome during pathogenic infections.
THE NLRP3 INFLAMMASOME AND HUMAN INFLAMMATORY DISEASES
NLPR3 was originally identified as the disease-responsible gene in patients with cryopyrinopathies, a group of rare autoinflammatory diseases that includes autosomal-dominant periodic fever syndromes, familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome, and chronic infantile neurological cutaneous and articular syndrome, which are collectively referred to as the cryopyrin/NLPR3-assocatied periodic syndromes (CAPS) [54,55]. Patients with gain-of-function mutations in and/or around the NLPR3 NACHT domain have primary symptoms, such as urticarial skin rashes and fever, in the absence of apparent infection [54,55]. The clinical manifestations of CAPS patients are caused by spontaneous secretion of IL-1β and IL-18, resulting from the constitutively active form of NLRP3 and continuous caspase-1 and NF-κB activation [6,56]. A variety of endogenous danger molecules have been reported in NLRP3-dependent molecular mechanisms of inflammatory disease pathogenesis. In this chapter, we discuss the roles of the NLRP3 inflammasome in inflammatory diseases, including gout, atherosclerosis, and type II diabetes (Fig. 2).
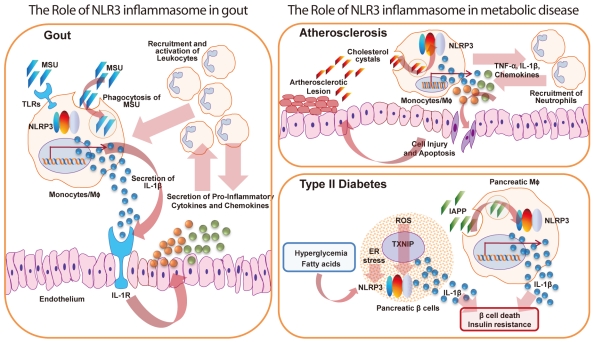
The NLR-related protein 3 (NLRP3) inflammasome in various inflammatory diseases. The NLPR3 inflammasome is involved in the pathogenesis of a variety of inflammatory diseases, including gout, type II diabetes (T2D), and atherosclerosis. In gout, crystalline or particulate NLRP3 activators such as monosodium urate (MSU) crystals are engulfed and then recognized by toll-like receptors (TLRs) (i.e., TLR2 or TLR4), leading to the release of pro-interleukin (IL)-1β. These activators can also be phagocytosed by macrophages, leading to lysosome rupture and NLRP3 inflammasome activation (see details in Fig. 1). Such NLRP3 activation causes the release of IL-1β. IL-1β released from macrophages activates IL-1 receptors on epithelial cells and resident macrophages, resulting in signal transduction and leading to the release of pro-inflammatory cytokines and chemokines. These cytokines/chemokines in turn recruit and activate leukocytes, amplifying the inflammatory positive-feedback loop. This inflammatory cascade may be the major cause of gout. In atherosclerosis, cholesterol crystals in atherosclerotic lesions activate the NLRP3 inflammasome in macrophages, leading to inflammation and cell infiltration. This amplified inflammatory cascade leads to the accumulation of extracellular lipids, resulting in cell injury and/or death and increased atherosclerosis progression. In T2D, hyperglycemia or fatty acids directly activate the NLRP3 inflammasome, causing the release of inflammatory cytokines and IL-1β. Prolonged hyperglycemia in pancreatic islets leads to reactive oxygen species (ROS) production and endoplasmic reticulum (ER) stress. Increased ROS levels trigger NLRP3 inflammasome activation through a thioredoxin-interacting protein (TXNIP)-dependent pathway. The elevated ER stress in β-cells also activates the NLRP3 inflammasome, but the reason and mechanism for this remain unknown. Islet amyloid polypeptide (IAPP) activates the NLRP3 inflammasome in pancreatic macrophages and results in the secretion of pro-inflammatory cytokines and IL-1β. The elevated levels of IL-1β from pancreatic macrophages and β-cells result in increased β-cell death and insulin tolerance, leading to reduced insulin production.
Gout and the Inflammasome
Gout is an autoimmune disorder with inflammatory arthritis caused by hyperuricemia that results in the deposition of MSU crystals in joints and soft tissues [57]. Martinon et al. [10] showed that MSU and calcium pyrophosphate dihydrate (CPPD) crystals activate the NLRP3 inflammasome, resulting in the production of active IL-1β and IL-18. Mice deficient in inflammasome components or the IL-1Rβ receptor have impaired neutrophil influx in an in vivo model of crystal-induced peritonitis. Recent studies of murine gout models [58,59] as well as clinical studies [60,61] have shown that IL-1β is involved in gout pathogenesis as a key proinflammatory cytokine that causes increased neutrophil influx into the synovium and joint fluid. In addition, in bleomycin-induced lung injury models, pulmonary accumulation of uric acid released from injured cells activates the NLRP3 inflammasome, leading to IL-1β production, in lung injury leading to pulmonary inflammation and fibrosis [62].
Atherosclerosis and the NLRP3 Inflammasome
Atherosclerosis is a progressive inflammatory disease and an underlying cause of cardiovascular disease, which is linked to certain risk factors, including dyslipidemia and hypertension [63,64]. Arterial wall injury is thought to be the results of an inflammatory process, based on the cell injury and apoptosis induced by cholesterol crystals deposited in the necrotic core of atherosclerotic plaque lesions [65,66]. During the vicious developmental cycle of atherosclerosis, macrophage inflammatory signals and accumulation of extracellular lipids lead to intimal injury [65,66]. Previous studies have shown that Chlamydia pneumonia, a pathogen associated with several chronic inflammatory conditions including atherosclerosis, can activate the release of IL-1β in unprimed bone marrow-derived macrophages and that the NLRP3/ASC complex is required for caspase-1 activation and maturation of IL-1β by this pathogen [67]. Moreover, IL-1R-deficient mice are more susceptible to C. pneumonia-induced pneumonia, suggesting a role for IL-1 in host defense against acute bacterial pneumonia caused by C. pneumonia [67].
Cholesterol crystals, common constituents of atherosclerotic lesions, activate the NLRP3 inflammasome by mechanisms involving potassium efflux and phago-lysosomal damage [68,69]. Thus, NLRP3 inflammasome-mediated IL-1β release may contribute to inflammatory processes during atherosclerosis and cell injury, driving disease progression. Future studies modulating IL-1β secretion and NLRP3 inflammasome activation will inform new strategies for controlling the potential initiation and progression of inflammation in atherosclerotic lesions.
Type II Diabetes and the NLRP3 Inflammasome
Type II diabetes (T2D) is characterized by a combination of insulin resistance and pancreatic β-cell dysfunction [70]. Numerous reports have indicated that obesity and T2D are associated with an increased systemic, proinflammatory milieu. Notably, IL-1β [71] and IL-18 [72] have been shown to risk factors for T2D. Particularly, IL-1β has profound cytotoxic effects on pancreatic β-cells and inhibitory functions on islets [73]. High glucose or free fatty acids, which are elevated in T2D, can directly induce inflammatory responses and increased IL-1β production [74]. Macrophage-derived IL-1β production in insulin-sensitive organs leads to progression of inflammation and induction of insulin resistance in obesity [75]. Many studies in animals and humans have reported that IL-1β-targeted therapy can be useful as an immunomodulatory strategy for T2D treatment with improved glycemia, β-cell functional mass, and insulin sensitivity, and can reduce inflammatory responses [76,77].
Islets of T2D patients have amyloid deposition and increased production of proinflammatory cytokines and chemokines. This unique, primary component of islet amyloid deposits is the islet amyloid polypeptide (IAPP; also known as amylin) [78]. Recent reports have shown that IAPP can directly induce the generation of IL-1β in macrophages through NLRP3 inflammasome activation [79]. In addition, human (h) IAPP, but not nonamyloidogenic rodent IAPP, induces islet chemokine secretion and production of a variety of proinflammatory cytokines from macrophages [80]. Treatment with the IL-1R antagonist (IL-1Ra) anakinra greatly improves glucose tolerance in non-obese diabetic/severe combined immunodeficient recipients of islet grafts from hIAPP-expressing transgenic mice, suggesting that amyloid-induced IL-1β secretion and inflammasome activation contributed to the β-cell dysfunction of islet transplantation [80].
The NLRP3 inflammasome is increasingly recognized as a metabolic sensor of metabolic dysregulation, such as T2D pathogenic status [13]. Triggering of inflammasome activation causes thioredoxin-interacting protein (TXNIP) to associate with NLRP3 in an ROS-dependent manner, leading to the subsequent secretion of IL-1β and insulin resistance [13,81]. In addition, Txnip(-/-) or Nlrp3(-/-) mice show improved glucose tolerance and insulin sensitivity [81]. However, another study showed that there is no difference in IL-1β secretion between wild-type and TXNIP-deficient macrophages in response to NLRP3 stimulation [79]. Although this discrepancy has not been fully clarified, one recent study has shown that glucose, but not fatty acids, induce islet β-cell expression of proapoptotic TXNIP [82]. This study also revealed that TXNIP deficiency efficiently protects against staurosporine- or glucose-induced β-cell toxicity; however, it is not as protective against endoplasmic reticulum (ER) stress- or fatty acid-mediated lipoapoptosis [82]. Future studies should clarify the exact role of TXNIP in terms of ROS-dependent inflammasome regulation, including additional investigation in different experimental settings.
T2D patients have an increased ER stress responses, which is often involved in the pathology of chronic inflammatory diseases [83]. Recent studies have shown that ER stress can activate the NLRP3 inflammasome, resulting in the subsequent release of IL-1β [84]. Interestingly, ER stress-induced NLRP3 inflammasome activation depends on ROS generation and potassium efflux, but not the classic unfolded protein response [84]. Further studies should reveal the underlying mechanism(s) by which the ER stress response contributes to chronic inflammatory disease through inflammasome activation.
Clinical Trials Testing Modulation of inflammasomes
Inflammasomes and IL-1β are increasingly recognized as drug targets for a variety of inflammatory disorders. Substantial advances in our understanding of the molecular mechanisms of inflammasome activation have resulted in clinical trials that have tested therapeutic strategies targeting IL-1 production. It is well known that IL-1 blockade with either the human IL-1β neutralizing monoclonal antibody canakinumab (Ilaris, Novartis Pharmaceuticals Co., Basel, Switzerland) or the IL-1 receptor antagonist (IL-1Ra) anakinra (Kineret, Amgen, Thousand Oaks, CA, USA) potently inhibit inflammatory effects and joint damage in rheumatoid arthritis [85] and have remarkable therapeutic effects on patients with Muckle-Wells syndrome [86]. IL-1Ra also ameliorates the clinical symptoms and hematological features of patients with familial cold autoinflammatory syndrome (FCAS), a disease with genetically defective NLRP3 [87]. In addition, rilonacept (Arcalyst, Regeneron Pharmaceuticals Inc., Tarrytown, NY, USA), a fusion protein of the ligand-binding domains of human IL-1R and IL-1R accessory protein (IL-1RAcP) with the Fc portion of human immunoglobulin G1, may improve the clinical symptoms and signs of patients with CAPS [88]. These findings seem to be promising and support the development of a new IL-1β-targeted therapy for more common inflammatory diseases.
Colchicine has been used for treatment of acute gouty attack [89]. Uric acid crystal-induced NLRP3 inflammasome protein complex assembly is inhibited by colchicines through microtubule inhibition [10], thus explaining its role upon activation of the NLRP3 inflammasome in gout pathogenesis. Clinical studies of the IL-1 inhibitors rilonacept (IL-1 Trap) [90], canakinumab (monoclonal anti-IL-1β antibody) [91] and anakinra [92] have shown therapeutic benefit for patients with acute and chronic gout. In addition, an oral caspase-1 inhibitor, VX-765 (Vertex Pharmaceuticals Inc., Cambridge, MA, USA), inhibits IL-1β secretion from LPS-stimulated peripheral blood mononuclear cells [93]. In contrast to IL-1β-targeted therapy, caspase-1 inhibitors are not as efficient, despite their good efficacy shown in experimental models [94]. Developing new agents may be important for patients with chronic, refractory, inflammatory conditions.
CONCLUDING REMARKS
Much progress has been made in understanding the molecular mechanisms of activation and roles of the NLRP3 inflammasome. However, numerous questions remain, including the molecular basis for NLRP3 recognition by unidentified ligands, its distinct role in the context of various physiological conditions, and the regulatory pathways governing NLRP3 inflammasome activation. Although the NLRP3 inflammasome has been identified as a critical immune defense platform in a variety of bacterial, fungal, and viral infections, the precise mechanisms by which it induces host immune activation have not been fully clarified. Moreover, many pathogens, especially those that invade the cytosolic environment, can escape, dysregulate, resist, or even co-opt use of the NLRP3 inflammasome complex. Because of their key roles in inflammatory disease, IL-1 and inflammasomes have been targets of recent clinical trials for inflammatory disease. As NLRP3 inflammasome activation also provides significant host defense functions, caution is needed with regard to the over-inhibition of IL-1 when therapeutically modifying host immunity to pathogenic infection. As the NLRP3 inflammasome is important not only in host defense but also in inflammatory disease pathogenesis, challenges remain for the development of novel targets and appropriate situation-dependent adjustments for the treatment of infectious and inflammatory disorders.
ACKNOWLEDGEMENTS
We are indebted to current and past members of laboratory for discussions and investigations that contributed to this article. We also thank Jae-Min Yuk and Hyo Sun Jin for critical reading of the paper. This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-0027459) and by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A100588).
Notes
No potential conflict of interest relevant to this article was reported.