Presynaptic Dysfunction by Familial Factors in Parkinson Disease
Article information

Abstract
Parkinson disease (PD) is the second most prevalent neurodegenerative disorder after Alzheimer disease. The loss of specific brain area, the substantia nigra pars compacta is known as a major etiology, however it is not fully understood how this neurodegeneration is initiated and what precisely causes this disease. As one aspect of pathophysiology for PD, synaptic dysfunction (synaptopathy) is thought to be an earlier appearance for neurodegeneration. In addition, some of the familial factors cumulatively exhibit that these factors such as α-synuclein, leucine-rich repeat kinase 2, parkin, PTEN-induced kinase 1, and DJ-1 are involved in the regulation of synaptic function and missense mutants of familial factors found in PD-patient show dysregulation of synaptic functions. In this review, we have discussed the physiological function of these genetic factors in presynaptic terminal and how dysregulation of presynaptic function by genetic factors might be related to the pathogenesis of Parkinson disease.
INTRODUCTION
Parkinson disease (PD) is the second most common neurodegenerative disorder after Alzheimer disease (AD). Its’ etiology is characterized by progressive decay of dopaminergic neurons in the substantia nigra pars compacta that cause depletion of dopamine in the brain and also the appearance of cytosolic aggregation known as Lewy bodies which are composed of α-synuclein, however the pathogenesis of PD still remains to be fully understood. Genetic researches for more than 2 decades in PD have allowed identifying about 2 dozen monogenic forms of genetic factors to develop PD such as SNCA, parkin, PINK1, LRRK2, and DJ-1 [1-7]. Furthermore, continuously these lists of PD-related genes are growing which might be able to provide a valuable mechanism of pathogenesis of the PD.
Synapse is a fundamental unit for neural communication, which is composed of pre- and postsynapse. In the aspect of the presynaptic terminal, it is essential for synaptic transmission upon neural activity. When action potential arrived at nerve terminal, voltage-gated Ca2+ channels are opened and subsequently, Ca2+ influx occurs into the cytosol of synapse. Synaptic vesicles which contain neurotransmitter in the lumen and various proteins in the synaptic vesicle membrane including a Ca2+ sensor (synaptotagmin I) and vesicle fusion machinery (SNARE complex) are localized in release area near to voltagegated Ca2+ channels. After sensing Ca2+, consequently, neurotransmitter is released in release area (active zone) by fusion of synaptic vesicle. During this process in the millisecond scale, postsynaptic area is ready to take a neurotransmitter signal with various receptors. Many receptors are localized at the surface membrane to take their appropriated ligand (e.g., N-methyl-D-aspartate receptor, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, metabotropic glutamate receptor, gamma-aminobutyric acid receptor). In accordance with this process, neuronal information collectively flows with efficient for a proper function of brain. Likewise, as a basic communication unit in the brain, presynaptic physiology is essential for initiation of communication and it is responsible for most upstream part of neural information flow. Regarding neural disease initiation, structural and functional dysregulation of synapse is one of the major causative factors for various neural diseases. Particularly, synaptic dysfunction is thought to be an earlier point for disorder [8,9], and neurodegenerative disorder such as PD is also thought to be synaptic dysfunction is the very early symptom. For example, exocytosis impairment of synaptic vesicle indicates the initial defects of progression for PD [10-12]. Interestingly, major prevalent familial factors of PD are mostly involved in presynaptic functions. Some of the PD-related proteins are dominantly localized at nerve terminals and associate with synaptic vesicle at nerve terminals, by doing so it modulates synaptic vesicle behavior such vesicle fusion and vesicle recycling [10,13]. Another group of genetic factors which have an enzymatic activity has many substrates among presynaptic proteins [14,15], implying that many genetic factors are related to the regulation of presynaptic function.
Since neural information flow is triggered in the presynaptic terminal by secretion of neurotransmitter, this review mainly discusses some of core genetic factors’ function in presynaptic terminals. How these familial factors control functional and structural aspect of presynaptic terminals such as release probability, synaptic vesicle recycling, synaptic vesicle size and functional synaptic vesicle pool mobility, and also a functional interaction of presynaptic subcellular organelle with synaptic regulation.
ALPHA-SYNUCLEIN IN PRESYNAPTIC TERMINAL
α-synuclein, a 15-kDa protein, is highly enriched in presynaptic terminals. Although it is still not much known what is the general function of α-synuclein at synapses, it has been related to synaptic vesicle trafficking and its dynamics because it associates with synaptic vesicles [16,17]. In the aspect of synaptic transmission, several studies have reported that α-synuclein controls neurotransmitter release in nerve terminals. The level of α-synuclein affects the efficacy of synaptic transmission, for example when the amount of α-synuclein is increased by exogenously expressing α-synuclein, synaptic transmission is decreased [10,18-20] and vesicle priming for ready to release is suppressed by α-synuclein overexpression in PC12 and chromaffin cells [21]. Alpha-synuclein specifically inhibits vesicle docking by binding acidic lipid-containing membranes [22]. By association with trafficking protein, Rab small GTPase, synaptic vesicle pool size is modulated [23]. For the positive influence of synaptic transmission, vesicle fusion pore formation is enhanced by α-synuclein [24]. A pathogenic mutant of α-synuclein also alters synaptic function. Expression of the pathogenic mutant form of α-synuclein, A53T in the calyx of Held synapse impairs synaptic transmission [25]. In the aspect of synaptic retrieval, α-synuclein is highly involved in synaptic vesicle endocytosis. First, α-synuclein is required for the fast kinetics of synaptic vesicle endocytosis [13] and induce clathrinmediated synaptic vesicle endocytosis [26]. Furthermore, the level of α-synuclein expression impairs synaptic vesicle reclustering after endocytosis [10], and acute treatment of human recombinant α-synuclein leads to impairment of synaptic vesicle endocytosis, and subsequently affects to synaptic transmission [27]. Interestingly Pathogenic mutant form of α-synuclein A53T but not A30P is involved in synaptic vesicle endocytosis [25,27]. The expression of A53T α-synuclein mutant in the genetically manipulated calyx of Held terminals impairs fast and slow endocytosis of synaptic vesicle and alters replenishment of readily releasable pool as well. Consistently, exogenous treatment of human recombinant α-synuclein mutant A53T impairs synaptic vesicle endocytosis at the immature calyx of Held terminals [27].
Structurally, α-synuclein also participates in presynaptic architecture [28]. Presynaptic terminal has a complex structure with synaptic vesicle and subcellular organelles. In general, nerve terminal of central nervous system neuron (e.g., a hippocampal pyramidal neuron) contains about 100–200 synaptic vesicles with–40 nm diameter of the mean [29]. Since α-synuclein associates with synaptic vesicle, in the absence of α-synuclein, the size of presynaptic terminal is collectively decreased. In addition, with pathogenic mutant A30P α-synuclein expression, synaptic vesicle distribution is severely altered in the proximal area of synaptic terminals.
Accordingly, α-synuclein is deeply involved in presynaptic functions by means of synaptic transmission and synaptic retrieval. Pathogenic mutant, A53T, has a structural and functional influence for the presynaptic terminal, and it is likely that α-synuclein is a proper target in synaptic pathogenesis for PD.
LRRK2 IN PRESYNAPTIC TERMINAL
Leucine-rich repeat kinase2 (LRRK2) is a large protein (286 kDa) that has multiple domains including, GTPase, Kinase, and several protein-protein interaction domains. Several missense point mutations of LRRK2 are identified and it is thought to be a major causative genetic factor of PD not only familial but also sporadic PD. However, it is still elusive what the normal function of LRRK2 is and how LRRK2’s mutations cause neurodegeneration. In the aspect of synaptic function, many of synaptic proteins are substrates for LRRK2, implying that it might implicate in synaptic function and its’ pathogenic mutants might dysregulate synaptic function. One of the most prevalent mutants, G2019S LRRK2, enhanced kinase activity form, is implicated in synaptic transmission. G2019S knock-in mice reveal that glutamate and dopamine neurotransmission is gradually decline in the course of aging [30]. Striatal spiny projection neurons in G2019S Knock-in mouse decrease excitatory postsynaptic current amplitude [31], and also it impairs synaptic plasticity [32]. However, some reports show a reverse phenotype that G2019S knock-in neurons exhibit the potentiation of synaptic transmission [33]. This conflict probably comes from a different method with no considerable age status. Another missense mutant, R1441C, also reveals similar synaptic defect. R1441C Knock-in mice initially show no dopaminergic neurodegeneration, however, after 2 years, dopamine D2 receptor function is altered with decreased locomotor activity implying that dopamine transmission and D2 receptor function are impaired [34].
In addition to regulation of synaptic transmission, LRRK2 has also an important function in synaptic retrieval process at presynaptic terminal, which is essential for maintenance of presynaptic functionality. The interaction of LRRK2 with Rab5 identified by yeast 2-hybrid screening modulates synaptic vesicle endocytosis by the cooperating process [35]. The ablation of LRRK2 impairs synaptic vesicle endocytosis in striatal neurons and synaptic vesicle number is significantly decreased [36,37] and neurons in ventral tegmentum area, mainly composed of dopaminergic neurons, are also impaired in synaptic vesicle endocytosis [38]. Furthermore, LRRK2 phosphorylates several endocytic proteins. Endophilin, one of the major endocytic proteins, is recently identified as a substrate for LRRK2 kinase. LRRK2 implicates in synaptic vesicle endocytosis by phosphorylating endophilin S75, which induces endophilin-mediated membrane association and tubulation, and LRRK2 G2019S expression exhibits a reduction of synaptic vesicle endocytosis [14,15]. In addition, phosphor-regulation of endophilin by LRRK2 provides a station for macroautophagy at presynaptic terminals [39,40]. It will be next question that how dysregulation of endocytosis by LRRK2 G2019S connect to the pathogenesis of PD and what other missense mutants of LRRK2s do affect in synaptic vesicle endocytosis.
PARKIN IN PRESYNAPTIC TERMINAL
Parkin is an E3 ubiquitin ligase that expressed in brain and various tissues. As a ubiquitin ligase enzyme, it has a number of substrates and is involved in various biological functions such as mitochondria stability, transcriptional regulation. Parkin also regulates a variety of cellular process and physiology, however precise function of parkin is still elusive. In the nervous system, parkin has been known that mutation of parkin gene is one of the causative factors for early-onset PD. As cumulating reports indicate that synaptic dysfunction by parkin might be related to the early symptom of pathogenesis for PD. Some synaptic proteins interact with parkin such as CDCrel-1, synaptotagmin. CDCrel-1-degradation induction by parkin affects synaptic dysfunction, which induces a decrease of synaptic transmission [41]. Without parkin protein, synaptic transmission is also impaired. Parkin knockout mice reveal that evoked dopamine transmission is reduced [42] as well as decrease glutamate transmission by impairing AMPAR endocytosis [43]. However, interestingly it has reported that spontaneous release of dopamine is reversely increased in parkin knockout mice [42]. Dopaminergic neurons derived from patient-based human-induced pluripotent stem cell exhibit that spontaneous release of dopamine is also increased [44], implying that the patient has somehow dominant negative parkin form. In the aspect of synaptic retrieval with parkin, no direct evidence is reported by far. However, interestingly, endophilin, one of the key proteins for endocytosis, knock out brain shows highly upregulation of parkin expression [45], implying that it might be also implicated in synaptic vesicle endocytosis. It will be next target whether parkin implicates in the synaptic retrieval process.
PINK1 IN PRESYNAPTIC TERMINAL
PTEN-induced kinase 1, PINK1, is a mitochondrial serine/threonine-protein kinase. It has been known that it controls mitochondria stability. By interacting with parkin, PINK1 regulates parkin activity and consequently modulates mitophagy. It is also known, mutation of PINK1 is a causative factor for early-onset familial PD. Striatal neurons in PINK1 knockout mice is altered electrical activities, and subsequent investigation for synaptic function reveals that dopaminergic neurons in PINK1 knockout mice display abnormal development and it leads to dysfunction of dopamine release which might be related to an early pathogenic mechanism of PD by PINK1 [46]. And PINK1 null mice also show that the expression of DA receptors is unchanged but evoked dopamine release and its quantal size are decreased [47]. However on the contrary enhanced excitatory transmission in PINK1 knockout neurons is also observed. Hippocampal neurons without PINK1 increase spontaneous glutamate release [48]. It is noted that PINK1 is implicated in synaptic function with variability upon neuron type and their activities (evoked and spontaneous). PINK1 is also an important mediator between mitochondria and presynaptic function. In the treatment of low-dose chronic rotenone, mitochondria functional inhibitor, in PINK1 deleted neurons significantly impact synaptic plasticity [49] and PINK1 mutant mice and fly exhibit defect of mitochondria complex I, which subsequently cause loss of dynamics of synaptic vesicle during rapid activity, and it is restored when additional ATP is supplied [50]. In the aspect of synaptic vesicle recycling, no related study is reported, implying that PINK1 may not be related to synaptic vesicle endocytosis.
DJ-1 IN PRESYNAPTIC TERMINAL
DJ-1 also known as park7 is small a 20-kDa protein. It is widely expressed in neuron and nonneuronal cells. It is known as a cellular sensor for oxidative stress and a redox-mediated chaperone, however precise function of DJ-1 still needs to be understood. Missense mutations of DJ-1 are one of the causative factors for familial PD. The role of DJ-1 in synapse has been studied. It has reported that DJ-1 associated with synaptic membrane, and localized in presynaptic terminals [51,52]. DJ-1 interacts with monoamine transporter (vMAT2) and controls the activity of vMAT2 which might consequently connect to the regulation of synaptic function [53]. It also modulates synaptic plasticity in hippocampal CA1 synapses [54]. In the aspect of synaptic vesicle trafficking, synaptic vesicle endocytosis is defected in DJ-1 ablated neurons. The rate of endocytic time constant is about two times slower than one of WT and it eventually affects in a decrease of neurotransmission during repetitive neural activities [52].
OTHERS IN PRESYNAPTIC TERMINAL
As a genemic sequencing technology is rapidly developing, recently new familial factors of PD have been being more identified. Based on genome-wide association study (GWAS) from PD patients, several new PARKINs members have been identified. The Sac1 domain mutation in synaptojanin1 is identified in early-onset progressive Parkinsonism along with seizure in Italian families [55,56]. This identification is continuously following in Indian family [57], although no mutation found in Taiwanese family [58]. Synaptojanin1, a lipid phosphatase, is known as one of key endocytic protein, which is responsible for synaptic vesicle endocytosis and reavailability by providing uncoating process of coated vesicle. Rescue with this missense mutation of synaptojanin1 impairs synaptic vesicle endocytosis and axonal structural change [59], implying presynaptic dysfunction by synaptojanin1 also is implicated in the pathogenic process of disease.
Another factor recently identified with several point mutations is transmembrane protein 230 or TMEM230. With GWAS and several sequencing analysis from North American families and Chinese cohort, 4 different mutations were identified [60]. It localizes subcellular organelle and secretory vesicles [61]. Because of this specific localization, it might be involved in vesicle trafficking. However, it still remains to be explored. Interestingly following up sequencing analysis from other various ethnic populations (e.g., Taiwanese, Han Chinese, and Japanese) reveal that significant evidence of mutation in TMEM230 has not been identified. Collectively, more study and analysis will be needed to address PD-relation of this gene.
Endophilin is not the PD related protein, however many of studies show that some PD-related factors functionally and genetically interact with endophilin such as LRRK2 and parkin. Although no direct evidence is reported regarding endophilin and PD, it might be indirectly associate with PD process.
CONCLUSIONS
PD is the second largest neurodegenerative disease with fast growth. Although the pathogenesis of PD is still not precisely understood, specific part of the brain (substantia nigra par compacta) degeneration and Lewy body accumulation with unknown reason are reported. In addition, recently many genetic mutations were identified from PD patients. Recently cumulating studies reveal that familial factors of PD such as α-synuclein, LRRK2, Parkin, PINK1, DJ-1, and others are implicated in synaptic physiology and mutants’ form of these factors result in dysregulation of synaptic physiology (Fig. 1). Alpha-synuclein modulates synaptic transmission and synaptic vesicle retrieval. LRRK2 phosphorylates endocytic proteins and synaptic proteins. By doing so, it controls presynaptic terminal physiology including synaptic vesicle trafficking. Parkin and PINK1 also modulate synaptic plasticity along with mitochondria integrity. DJ-1 participates in synaptic vesicle endocytosis and reavailability. Synaptojanin1, TMEM230 have just joined as a PD-related factor although more study is needed. These synaptic dysfunctions are not a terminal symptom but the very early process of neural diseases. Therefore more details of understanding how these factors alter presynaptic physiology will give an understanding of early pathogenesis and appropriate strategy for finding a therapeutic target for early intervention of PD.
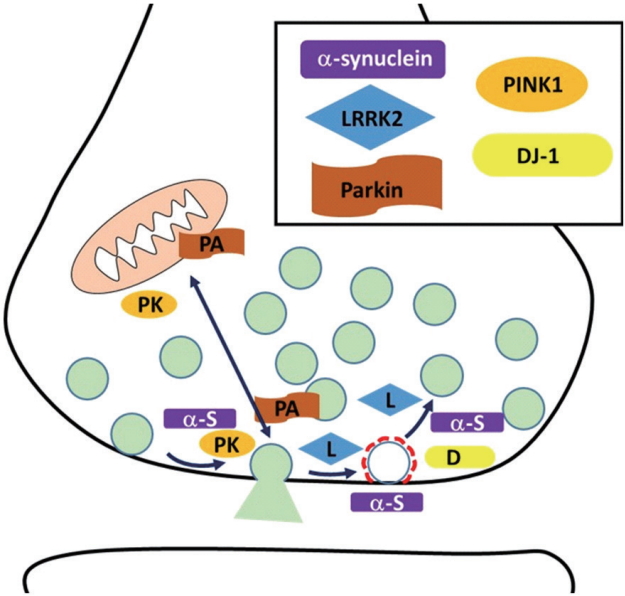
Schematic display of familial factors of Parkinson disease (PD) in presynaptic terminal. The familial factors of PD, α-synuclein, leucine-rich repeat kinase 2 (LRRK2), parkin, PTEN-induced kinase 1 (PINK1), and DJ-1 are localized at presynaptic terminal. Alpha-synuclein, parkin (PK), and PINK1 (PA) regulate synaptic transmission. Alpha-synuclein, LRRK2 (L), and DJ-1 participate in synaptic vesicle endocytosis.
Acknowledgements
I would like to thank members of SHK laboratory for the valuable discussion and comments.
Notes
Fund/Grant Support
This work is supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science and ICT (NRF-2017R1A2B4007019) and a grant from Kyung Hee University (KHU-20160702).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: SHK
·Study concept and design: WL, SHK
·Acquisition of data: WL, SK, SH
·Drafting of the manuscript: WL, SHK
·Critical revision of the manuscript for important intellectual content: SHK
·Obtained funding: SHK
·Administrative, technical, or material support: WL, SK, SH
·Study supervision: SHK