INTRODUCTION
Lower urinary tract symptoms (LUTS), characterized by an increased frequency of micturition, urgency, urge incontinence, and urinary obstruction, are very common in the geriatric population, a group that is rapidly increasing in number [1-3]. Antimuscarinic agents are widely used as first-line therapy for LUTS due to benign prostatic hyperplasia and overactive bladder (OAB) [4,5], because parasympathetic innervation is the predominant stimulus for bladder contraction [2]. These agents are also associated with anticholinergic side effects, however, including dry mouth, constipation, somnolence, and blurred vision, because the muscarinic receptor mediates the excitatory and inhibitory actions of acetylcholine in the central and peripheral nervous systems [3]. Dry mouth is the most common of these complaints and decreases quality of life. Therefore, numerous studies involving antimuscarinic agents to treat OAB have focused on targeting the urinary bladder over the salivary gland. The incidence of side effects on the central nervous system (CNS) is generally lower than the incidence of dry mouth, but such effects may be of great concern in elderly patients because of an increase of blood-brain barrier permeability with aging [4,5]. In this respect, clinical studies have demonstrated increased cognitive sensitivity to scopolamine [6,7] and a reduced density of brain muscarinic receptors in the elderly [8]. Accordingly, it is important to evaluate the bladder selectivity of antimuscarinic agents used to treat OAB for optimal medication.
IN VIVO DRUG-RECEPTOR BINDING
Several clinically useful drugs targeting receptors for neurotransmitters and hormones are available. The chain of events from drug administration to a certain pharmacological endpoint is complicated but can be schematically simplified as illustrated in Fig. 1. It is evident that pharmacological effects of drugs are determined by both pharmacokinetic and pharmacodynamic processes. Pharmacokinetics includes the absorption, distribution, excretion, and metabolism of drugs, whereas pharmacodynamics includes the affinity of drugs for receptors, signal transduction, and homeostatic mechanisms [9,10]. The drug-receptor interaction results in a measured effect, and the magnitude of the interaction depends on the concentration of the drug in the biophase and further on its affinity for the receptors. The biophase concentration depends not only on the amount of drug given, but also on factors such as the pharmacokinetic processes. The in vivo pharmacological specificity may be complicated by several factors, including an equilibrium delay in concentrations of drugs between blood and the vicinity of receptors, the formation of active metabolites, the occurrence of acute tolerance or sensitization, and the involvement of physiological control mechanisms [11]. Thus, it may be important to examine directly the extracellular concentration, receptor occupancy, and pharmacological responses of drugs in different tissues such as target tissues and non-target tissues under in vivo conditions.
Currently, a number of novel drugs exhibiting target organ specificity, receptor subtype selectivity, and a long duration of action have been developed to reduce side effects and improve patient compliance [12-16]. The affinity of compounds for various receptors in the development of novel drugs has been evaluated mainly by using in vitro radioligand binding assays in tissue membrane preparations and in intact cells [13,15,17-20]. However, even if a compound displays high affinity towards a certain receptor in vitro, it is not necessarily able to reach such binding sites under in vivo conditions. In other words, the in vitro receptor binding characteristics may not necessarily ensure pharmacological specificity in vivo because various pharmacokinetic and pharmacodynamic factors are not taken into account. Therefore, techniques to measure in vivo receptor binding would be useful in clarifying the pharmacological specificity of drugs in relation to their pharmacokinetics. In this article, we describe the in vivo receptor binding characteristics in the bladder and other tissues of the antimuscarinic agents used to treat OAB. Furthermore, we discuss the rationale for the use of in vivo analysis of receptor occupancy in drug development.
SELECTIVITY OF BLADDER MUSCARINIC RECEPTOR BINDING
Transdermal Oxybutynin
Oxybutynin has been widely used to treat OAB, but its use is often limited by systemic side-effects such as dry mouth, blurred vision, constipation, and tachycardia, which appear frequently in patients receiving oral oxybutynin [21]. It has been shown that the controlled-release dosage form of oxybutynin (osmotic-controlled release oral delivery system, OROS oxybutynin) causes dry mouth less often than does the immediate-release form in patients with OAB [22,23]. It is also worth noting that a transdermal therapeutic system was shown to significantly improve the anticholinergic adverse effects of oral oxybutynin in patients with OAB [24,25]. In the novel dosage form of oxybutynin, a low plasma concentration of its active metabolite, N-desethyl-oxybutynin (DEOB), has been suggested to contribute to the low incidence of anticholinergic side effects [24,25].
Both oral and transdermal administration of oxybutynin result in a significant binding to muscarinic receptors in rat tissues, but a remarkable difference is seen between the oral and transdermal routes of administration in the effect on muscarinic receptors in the submaxillary gland [26]. Namely, although oral oxybutynin significantly decreases the maximal number of [3H]N-methylscopolamine (NMS) binding sites (Bmax) in the rat submaxillary gland and heart in a sustained manner, transdermal application of oxybutynin produces little reduction in Bmax values for [3H]NMS binding in rat tissue. On the basis of a kinetic analysis of binding parameters for radioligand by Yamada et al. [27], it is suggested that oral but not transdermal oxybutynin produces an extremely long-lasting (noncompetitive) blockade of muscarinic receptor sites in the submaxillary gland and heart, and that this might be due to a slowly dissociating blockade by oral oxybutynin of muscarinic receptors. Furthermore, in dose-response curves of pilocarpine-induced salivation, the antagonism by oral oxybutynin, unlike transdermal oxybutynin, was not simply competitive in that it suppressed the markedly maximal response by pilocarpine. Therefore, it is possible that a significant difference in exocrine muscarinic receptor binding characteristics under in vivo conditions underlies the partly lower incidence of severe dry mouth with transdermal rather than oral oxybutynin in patients with OAB [24,25].
Orally administered oxybutynin is rapidly absorbed from the gastrointestinal tract, with the major pathway of elimination by hepatic metabolism [28], and the plasma concentration of DEOB in humans after oral administration is much higher than that of oxybutynin [29]. On the basis of previous reports showing that the muscarinic receptor binding affinity of DEOB is greater in the salivary gland than in the bladder [30,31], it is considered that DEOB may be responsible for the long-lasting occupation of exocrine muscarinic receptors after oral oxybutynin. In fact, a similar concentration of DEOB to oxybutynin was detected after oral oxybutynin treatment in rats, but little of this metabolite was detected after transdermal oxybutynin treatment. Thus, it is possible that pharmacokinetic characteristics such as substantially less fluctuation in the plasma oxybutynin level and avoidance of a first-pass effect by transdermal oxybutynin brings about the significant difference in exocrine muscarinic receptor binding characteristics from that by oral oxybutynin, leading to the advantage of transdermal over oral oxybutynin in the treatment of OAB owing to a lower incidence of dry mouth.
In conclusion, transdermal oxybutynin may lead to a significant degree of binding to bladder muscarinic receptors without causing long-lasting occupation of muscarinic receptors in the submaxillary gland of rats and without the abolishment of salivation evoked by oral oxybutynin.
Solifenacin
In vitro radioligand studies with human recombinant muscarinic subtypes have shown that solifenacin exhibits high affinity and specificity for the muscarinic M3 subtype, mainly mediating contraction of detrusor smooth muscle, relative to the M1 and M2 subtypes [32]. In vivo studies in anesthetized rats have shown that solifenacin is 4 to 7 times more potent in inhibiting bladder contraction than salivation, whereas oxybutynin had little bladder selectivity [32,33].
After oral administration of solifenacin and oxybutynin, some difference was seen between the two drugs in the time course of the increases in dissociation constant (Kd
) for [3H]NMS binding in mouse tissues [34]. The increase in K
d with solifenacin in most tissues was greatest at 2 hours and was maintained for at least 6 or 12 hours, whereas the increase in K
d in each tissue reached a maximum at 0.5 hour after the oral administration of oxybutynin, followed by a rapid decline. This apparent distinction between solifenacin and oxybutynin appears to largely depend on their rate of increase and disappearance in the plasma. On the basis of the intensity and duration of the increases in K
d values, the muscarinic receptor binding activity of oral solifenacin in mice was suggested to be greatest in the submaxillary gland and lowest in the heart and also to be long-lasting in the bladder, prostate, submaxillary gland, and colon, in contrast with the transient binding in the heart and lung. Considering the subtype expression of muscarinic receptors in rat tissues [35,36], the observed selectivity of oral solifenacin may be interpreted to reflect the muscarinic subtype selectivity shown in the in vitro assay [32]. Additionally, the intensity and time-course of the inhibitory effects of solifenacin and oxybutynin on salivary secretion after oral administration seem to coincide reasonably well with those for muscarinic receptor binding in the submaxillary gland. Thus, oral solifenacin shows significant binding to muscarinic receptors in various tissues of mice, including the bladder, and the receptor binding activity of this agent may be long-lasting in most tissues expressing the M3 subtype [34,37].
Tolterodine
Tolterodine has been developed as a muscarinic receptor antagonist to treat OAB [38], and pharmacological and radioligand binding studies have shown that tolterodine exerts a potent antimuscarinic effect in the isolated detrusor muscle of guinea pigs and humans [39]. In addition, tolterodine has been demonstrated to display favorable tissue selectivity for the urinary bladder compared with the salivary glands in cats [39]. Tolterodine is extensively metabolized in the liver to form an active metabolite, 5-hydroxymethyl metabolite (5-HM) [40,41]. In in vitro experiments, tolterodine and 5-HM competed in a concentration-dependent manner with [3H]NMS for binding sites in the bladder, submaxillary gland, and heart of mice, and the potency of both agents was considerably greater than that of oxybutynin [42].
A significant difference seems to exist between oxybutynin and tolterodine in ex vivo muscarinic receptor binding characteristics in mouse tissues [42]. Oral administration of tolterodine compared with oxybutynin results in relatively slower and longer-lasting binding in each tissue, as characterized by the increase of K
d values for [3H]NMS, which is greatest at 2 hours and lasts for at least 6 or 12 hours. It should be noted that such extremely high receptor binding activity in the submaxillary gland as seen after oral oxybutynin is not observed with oral tolterodine and that the muscarinic receptor binding activity of oral tolterodine is of longer duration in the bladder than in the submaxillary gland. Furthermore, significant receptor binding activity by a lower dose of tolterodine is also observed in the bladder but not in the submaxillary gland. Therefore, oral tolterodine, unlike oral oxybutynin, binds more selectively to muscarinic receptors in the bladder than in the submaxillary gland. At these pharmacological doses, tolterodine is significantly weaker than oxybutynin in inhibiting pilocarpine-induced salivation of mice [42]. Although the mechanism by which oral tolterodine causes bladder receptor selectivity in mice is not clear, it has been shown that most of the administered dose in mice receiving oral [14C]tolterodine is preferentially distributed to the eliminating organs, that is, gall bladder, urinary bladder, liver, kidney, and lung [43]. Such high concentrations of tolterodine and 5-HM in the bladder may be attributable to the tissue selectivity of this drug.
In conclusion, oral tolterodine binds significantly to muscarinic receptors in the mouse bladder, and the receptor binding activity of this drug compared with oral oxybutynin is relatively slow and longer lasting.
Imidafenacin
Imidafenacin, a novel antimuscarinic drug with high tissue selectivity (organ specificity) for urinary bladder, was approved for treating OAB in Japan in 2007. Analysis of subtype-selectivity using recombinant human muscarinic receptors demonstrated that imidafenacin exerts higher affinity for the M1 and M3 than the M2 subtype, with inhibition constant (K
i) values in the low nM range [44]. Similar high potency and high selectivity for M1 and M3 over the M2 subtype was shown in the in vitro functional antagonism assays in isolated animal organs [44].
In the experiments in vitro, imidafenacin competed in a concentration-dependent manner with [3H]NMS for binding sites in the bladder, submaxillary gland, colon, lung, and brain, with a potency equal to or greater than that of oxybutynin [45]. Also, the affinity of imidafenacin for muscarinic receptors was significantly lower in the bladder than in the submaxillary gland or colon, whereas it was significantly greater than that in the heart. Greater affinity of imidafenacin for muscarinic receptors in the exocrine gland than in the bladder has also been observed in human tissues [46]. Because imidafenacin exhibits greater selectivity for the M3 than the M2 subtype [44], the high affinity of this drug for muscarinic receptors in the rat submaxillary gland reflects M3 subtype selectivity.
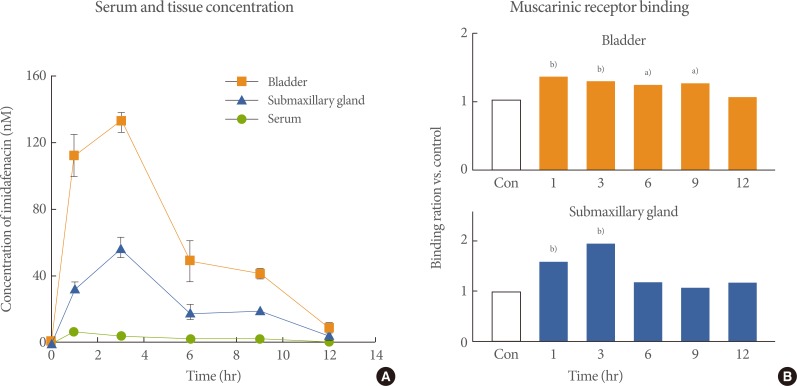
In a pharmacokinetic study in rats, the orally administered imidafenacin was distributed more to the bladder and submaxillary gland than to the serum, and the tissue concentration was much higher in the bladder than in the submaxillary gland (Fig. 2A) [45]. Orally administered imidafenacin was shown to bind muscarinic receptors more selectively in the bladder than in other tissues (Fig. 2B) [45]. The specific distribution of imidafenacin to the bladder might be related to its significant excretion into the urine. Up to 48 hours after the oral administration of imidafenacin (0.1 mg) in healthy volunteers, approximately 7.3% of the dose is excreted into urine as the parent compound and the maximum concentration is 293 nM [47]. Thus, imidafenacin may be transferred directly from urine to the bladder tissue by simple diffusion, and thus this agent could contribute greatly to the selective and long-lasting binding of bladder muscarinic receptors in rats. It can therefore be presumed that the significant binding of bladder muscarinic receptors by the excreted urinary imidafenacin is pharmacologically relevant in terms of the functional blockade of these receptors.
MUSCARINIC RECEPTOR SELECTIVITY IN THE BLADDER OVER THE BRAIN AS EVALUATED BY IN VIVO AUTORADIOGRAPHY
Muscarinic receptors play important roles in numerous physiological functions including higher cognitive processes such as memory and learning [48]. Deterioration in cognitive function during treatment with antimuscarinic drugs for OAB is of growing concern, particularly in elderly and dementia patients. In an autoradiographic study, the intravenous injection of oxybutynin, tolterodine, and solifenacin significantly decreased in vivo specific (+)N-[11C]methyl-3-piperidyl benzilate ([11C](+)3-MPB, a selective radioligand of muscarinic receptor) binding in each brain region of rats in a dose-dependent manner [49]. According to the estimated 50% receptor occupancy (RO50) values, the potency of muscarinic receptor occupancy by each agent in the rat brain appeared to be greatest for oxybutynin, followed by tolterodine and solifenacin. In contrast, darifenacin at pharmacologically effective doses did not significantly reduce in vivo specific [11C](+) 3-MPB binding so that the RO50 value could not be estimated. The RO50 values of antimuscarinic agents are similar to the intravenous doses inhibiting learning and memory behavior in rats [50]. Thus, these values could be regarded as the index of CNS pharmacological effects following the blockade of brain muscarinic receptors. Furthermore, the dose ratios (RO50/ID50) of antimuscarinic agents for the brain receptor occupancy to the inhibitory potency of increases in intravesical pressure are considered to reflect in vivo pharmacological selectivity for the urinary bladder over the brain. This ratio is relatively large for solifenacin (8.1 to 46.7) and tolterodine (3.6 to 17.9) compared with oxybutynin (1.4 to 3.4). Thus, the selectivity for the urinary bladder over the brain is relatively low for oxybutynin, suggesting a high feasibility of CNS side effects at the pharmacological doses used to treat OAB. The selectivity for the urinary bladder over the brain of solifenacin and tolterodine is clearly higher than that of oxybutynin in rats. In clinical studies, the incidence of CNS side effects of tolterodine was shown to be lower than that of oxybutynin and comparable to that of placebo [51-53]. Thus, these data suggest that solifenacin and tolterodine have advantages in the treatment of OAB owing to fewer CNS effects.
Antimuscarinic agents must first pass the blood-brain barrier to occupy central muscarinic receptors. The observed difference among antimuscarinic agents in the potency of brain muscarinic receptor occupancy may be defined by blood-brain barrier permeability, which is responsible for CNS effects in patients. The passive penetration of drugs through this physiologic barrier generally depends on physicochemical factors such as high lipophilicity, low degree of ionization (neutral charge), and small molecular size [51]. The characteristics of the chemical properties of oxybutynin relative to tolterodine, e.g., lipophilicity (log Ko/w: 4.68 vs. 1.83) [54,55] and neutral polarity (pKa: 6.44 vs. 9.87) [55,56], make it the most likely to cross the blood-brain barrier [57]. Additionally, the muscarinic receptor subtype selectivity of antimuscarinic agents may be implicated in the appearance of CNS effects. All five muscarinic receptor subtypes are expressed in the brain [35,58]. The M1 receptor is abundant in the cortex and hippocampus. In the striatum, the M1 and M4 receptors are distributed. By contrast, the M2 receptor is predominantly localized to the brainstem and cerebellum. The M3 receptor displays lower density in the brain compared with the M1, M2, and M4 receptors. The cognitive dysfunction by antimuscarinic agents may be mediated mainly by the M1 and M2 receptors in the CNS [59]. Oxybutynin shows selectivity for the M1, M3, and M4 receptors, whereas tolterodine and propiverine are relatively nonselective to muscarinic receptor subtypes [60,61]. Solifenacin and darifenacin show higher selectivity to the M3 receptor than to the M1, M2, and M4 receptors. Thus, the M1 selectivity in addition to the high blood-brain barrier permeability of oxybutynin may be more apt to cause CNS side effects. Such muscarinic receptor subtype selectivity may be partly associated with an observed difference among antimuscarinic agents in the in vivo potency of brain receptor occupancy.
BRAIN MUSCARINIC RECEPTOR BINDING EVALUATED BY POSITRON EMISSION TOMOGRAPHY
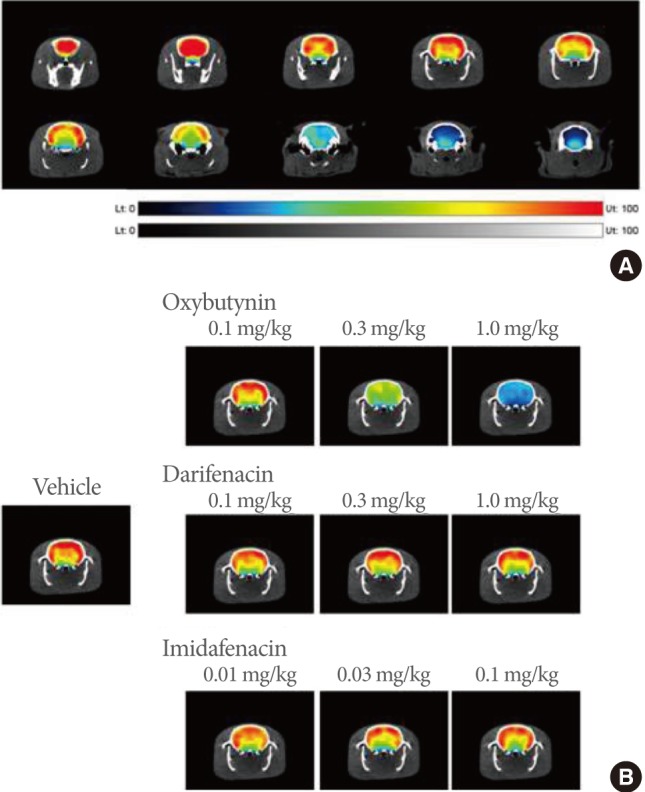
The in vivo imaging of brain receptors by positron emission tomography (PET) allows the precise localization of muscarinic receptors and the pharmacological characterization of muscarinic receptor antagonists [49,62]. Yoshida et al. [63] noninvasively characterized muscarinic receptor occupancy in rat brain after the systemic injection of imidafanacin, oxybutynin, and darifenacin by PET. The intravenous injection of oxybutynin but not imidafenacin or darifenacin at pharmacological doses decreased the binding potential of [11C](+)3-MPB in the cerebral cortex and corpus striatum in a dose-dependent manner (Fig. 3) [63]. The effects of imidafenacin and oxybutynin on the central muscarinic receptors and cognitive function in conscious monkeys were further investigated [64]. The occupancy levels of central muscarinic receptors and cognitive function were assessed with [11C](+)3-MPB-PET measurements and the titration version of the DMS (the delayed matching to sample) task (T-DMS task), respectively, in the same animals. Oxybutynin administered orally occupied muscarinic receptors dose-dependently 1 hour after administration, and cognitive function was significantly impaired in a dose-dependent manner. Imidafenacin also occupied muscarinic receptors to some extent, but did not induce cognitive impairment at all. This is consistent with pharmacological data showing that imidafenacin does not affect escape latency in the Morris water maze task in rats (spatial learning and memory) [65] and with clinical data showing that this agent is well-tolerated with fewer adverse effects [66-69]. Both imidafenacin and darifenacin have moderate polarity and low lipophilicity, suggestive of lower permeability. In addition, darifenacin is considered a substrate of P-glycoprotein, an active-transport system that carries this agent back across the blood-brain barrier [70].
CONCLUSIONS
The determination of in vivo drug-receptor binding has been shown to be useful in predicting the dose, potency, and duration of pharmacological effects of receptor antagonists. The analysis of muscarinic receptor binding characteristics in the bladder and other tissues after the systemic administration of antimuscarinic agents has revealed that in the treatment of OAB, systemic adverse effects such as dry mouth and cognitive dysfunction could be avoided by the new generation of antimuscarinic agents with high bladder selectivity. Consequently, the in vivo measurement of receptor occupancy by drugs may allow evaluation of pharmacological specificity from the integrated viewpoint of pharmacokinetics and pharmacodynamics.