What is the Cause of Recurrent Urinary Tract Infection? Contemporary Microscopic Concepts of Pathophysiology
Article information

Abstract
Urinary tract infections (UTIs) are the most common infectious disease and are mainly caused by Escherichia coli. In this review, we introduce the current concept of recurrent UTI (rUTI) based on recent research dealing with pathophysiology of the disease. Although urine is considered sterile, recent studies dealing with microbiome have proposed different ideas. UTIs have typically been considered as extracellular infections, but recently, uropathogenic Escherichia coli (UPEC) has been shown to bind and replicate in the urothelium to make intracellular bacterial communities. Binding UPECs might proceed in many ways including extracellular expulsion for clearance or survival and quiescent intracellular reservoirs that can cause rUTI. Moreover, it is also suggested that other important factors, such as lipopolysaccharide and multimicrobial infection, can be the cause of rUTI. This review article reveals a key mechanism of recurrence and discusses what makes a pathway of resolution or recurrence in a host after initial infection.
INTRODUCTION
Urinary tract infections (UTIs) are among the most common bacterial infections and are a significant problem worldwide. The most important issue is recurrence, whereby about 25% of women with UTI usually have recurrent UTI (rUTI) within 6 months after initial event. Many clinicians mainly use the definition of a rUTI as recurrence of 3 or more times in 1 year or more than 2 times in 6 months. It is reported that in about 25% of women the first UTI will have a second infection, and 25% of them will show a third and further recurrences [1]. Women are much more likely to show rUTI than men because of the risks involved with, sexual intercourse, urogenital ageing, pelvic organ prolapse, urethral diverticula, vesicovaginal fistula, urinary incontinence, menopause, and pregnancy. Moreover, there are urologic abnormalities that cause bacterial persistence, such as infectious urinary tract stones, bladder outlet obstruction, neurogenic bladder with high bladder pressure, and long-term indwelling of urethral or ureteral catheters [2].
The diagnosis of UTI is set up on detection in the urine of cells that are considered the most common causative factors, Escherichia coli, Proteus mirabilis, Klebsiella spp., and Group B Streptococcus (GBS) in pregnant women are also associated with UTI. The presence of the cells at concentrations ≥105 colony-forming unit/mL in urine has been considered as clinically significant bacteriuria [3]; however, the minimum number required to induce symptoms is undefined.
Recurrence of UTI is closely associated with antibiotic resistance. Emerging antibiotic resistance magnifies critical problems [4]. Increased use of antibiotics worldwide and prolonged use of prophylactic antibiotics or frequent use of several other antibiotics in patients with rUTI can contribute to the make of resistant bacteria; the event could develop within a few weeks. E. coli, which is resistant to at least 4 antibiotics, has been identified in fecal isolates from more than 90% of patients treated with prophylactic low-dose antibiotics for 2 weeks. Extended‐spectrum β‐lactamase‐producing bacteria, carbapenemase-resistant organisms, and pan‐resistant bacteria are serious problems [5].
The formation of intracellular bacterial communities (IBCs) in bacterial survival, pathogen recognition, and barrier function after antibiotic treatment, can play a critical role in the failure to eradicate pathogens [6-8]. The increase of antibiotic-resistant bacteria can make a serious problem of making rUTIs difficult to treat. Finally, clinicians cannot cure rUTI effectively, and it can cause a significant burden on health care systems. Even though antibiotic treatment has been successful in the treatment of acute UTIs, recent increases in the prevalence of antibiotic-resistant uropathogenic strains in the community have caused chronic and recurrent UTIs to become a common threatening disease [9,10].
The pathophysiology of rUTI is not well understood. By fully defining the risk factors contributing to the recurrence of uropathogenic E. coli (UPEC), the problem can be solved. The identification of host response and signal pathway that promote adherence and invasion of UPEC may explain the susceptibility of patients with rUTIs. However, it is not known how the molecular basis and adaptive immunity of rUTI are related to mechanisms of this disease. Here, we review the recent concepts of pathophysiology that may develop into rUTI.
CURRENT CONCEPTS OF PATHOPHYSIOLOGY IN RECURRENT URINARY TRACT INFECTION
Urinary Microbiota
The ‘microbiota’ shows microorganisms living inside an individual, however microbiome refers to the stack of genomes, genes, and products of the microbes [11]. Urine has been widely accepted as sterile in healthy human because of technical complexity in identifying the full scope of bacterial species. However, advances in technology and molecular biology have allowed detection of many microbiomes [12,13].
One research that assessed standard urine culture with next-generation sequencing of DNA reported that all patients who showed symptoms had positive results for urinary bacteria, however just 30% had positive results using standard culture [14,15]. The isolation of a specific bacteria in the urine can demonstrate critical implications in the development of certain diseases. Previous studies usually included standard urine cultures, which had important limitations for detection of the full set of bacterial species. New technique of expanded quantitative urine culture (EQUC) has made rapid development [16]. The negative results from urine cultures may attribute to the possibility that the patients have symptoms of UTI due to anaerobic bacteria or multimicrobial infection, rather than the only UPEC routinely detected for on standard urine culture [17-19]. Price et al. [20] revealed that standard method of urine culture missed about 90% of non-UPEC used by EQUC (Fig. 1).
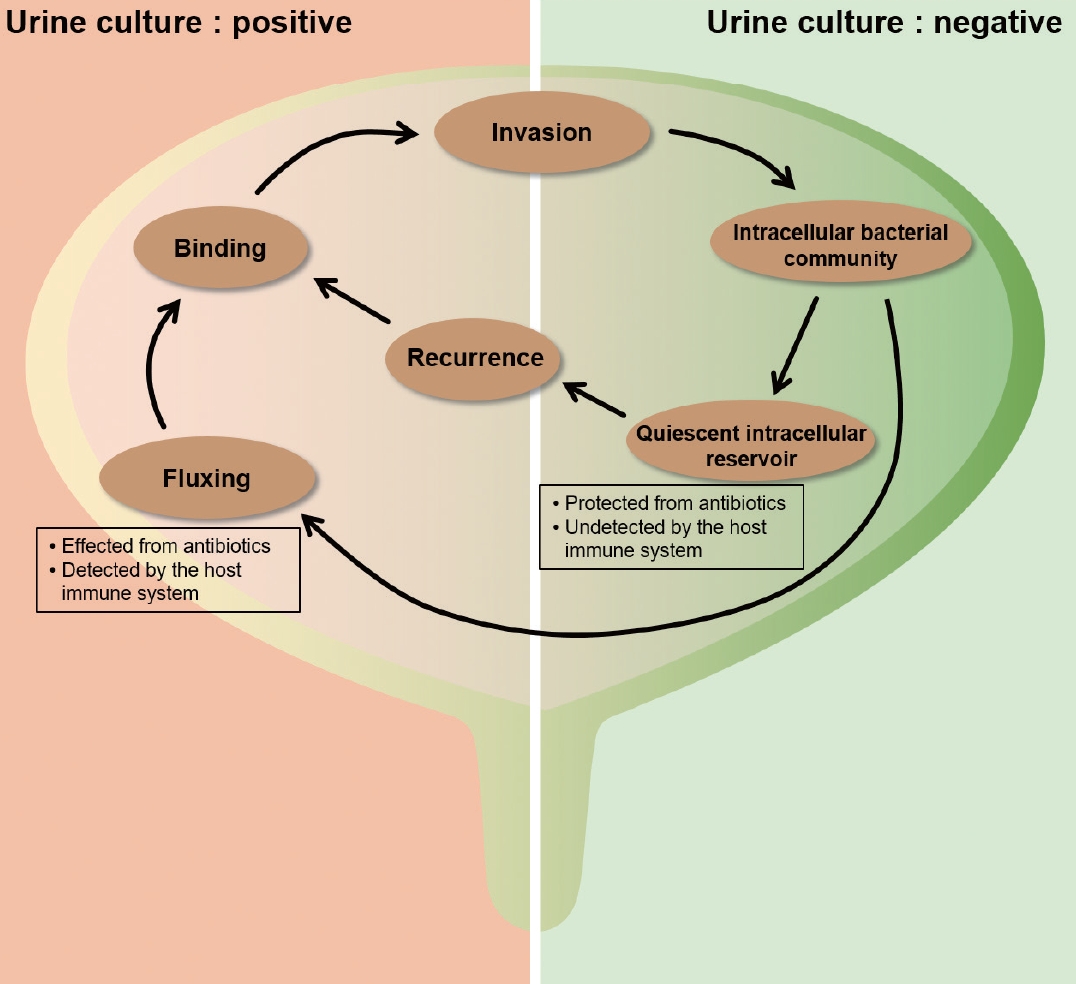
Diagram including flow chart of intracellular bacterial community (IBC) maturation and development. The left half (urine culture: positive) of the bladder shows phases of IBC development during which bacteria are in the intraluminal state in the bladder with bacteria susceptible to antibiotics and detected by the host immune system. The right half (urine culture: negative) of the bladder shows phases of IBC development during bacteria protected from antibiotic therapy and undetected by the host immune system.
It is usually considered that pathogens living in the gut are the origin of UTIs via periurethral contamination. Infections of the periurethra by a gut pathogen can be considered the starting point, followed by colonization in the bladder. However, data to support the hypothesis is limited. Indeed, a recent research showed that women with UPEC had different E. coli strains in their urine and stool [21]. Data dealing with the characteristics of pathogenic genes in increasing the trend for gut E. coli strains to induce UTI is also limited [22]. In contrast, another study has demonstrated the importance of pili and flagella in the invasion process of UPEC [23].
Spaulding et al. [24] reported the critical mechanism of UPEC persistence in the gut and recurrence of UTI. They demonstrated that F17-like and type 1 pili promoted intestinal colonization and bound to epithelial cells distributed along colonic crypts using a mouse model. Their results showed that F17-like pili are closely related to pilus types and restricted to extra-intestinal UPEC. After using high-affinity inhibitor of FimH, authors could see reduction intestinal colonization of UPEC without disrupting diversity of the gut microbiota.
A few studies have researched the starting step and the roles of gut microbiota in development of UTI. One research dealing with transplant recipients performed profiling of gut microbiota, and they reported an important relationship between gut Enterococcus abundance and Enterococcus UTIs [25]. The study investigated if the microbiota is a risk factor for induction of UTI and reported the important link between abundance of E. coli and recurrence of UTI. They showed that the gut abundance of Escherichia was associated with symptomatic Escherichia UTI development. Another study also proposed that increased fecal abundance of Escherichia and Enterococcus was involved with an increased risk of UTI [26].
Growing data support that microbiota can play an important role in the initiation of lower urinary tract symptoms (LUTS) [27]. Chen et al. [28] reported that UTI was common in patients with LUTS. Incontinence patients experienced rUTI with the same strain support the existence of a chronic reservoir, with the source. In experimental mice, E. coli can replicate in urothelium as IBCs and survive in a quiescent nonreplicating state [29]. Data for the existence of IBCs and quiescent bacteria has been reported in patients [30], supporting the hypothesis that the bladder may serve as a reservoir for recurrence. Another study also showed evidence of the presence of long-term reservoirs in the urothelium lining the urinary tract [17]. Furthermore, it has been shown that UPEC can form biofilms that are made of polysaccharide matrix encased in an uroplakin covering. The biofilms can induce recurrence as they protect bacteria from antibiotics and immune mechanisms.
There are growing data of a direct role for transient vaginal microbiota exposure in rUTI pathogenesis due to the vagina is a key anatomical site in the pathogenesis of rUTI in women. Vagina in women could serve as potential reservoir for UPEC. One important systematic review demonstrated that a Lactobacillus-dominated vaginal microbiota is correlated with health vaginal microenvironments [31]. Women who had decreased amounts of lactobacilli were found to be at high risk for a variety of urogenital disease, such as Neisseria gonorrhoeae, human immunodeficiency virus infection, and vaginal colonization with UPEC [32]. Gilbert et al. [33] studied that the role of vaginal bacteria exposure on the bladder using animal model. The authors and many scientists believed that rUTI might be triggered by Gardnerella vaginalis because G. vaginalis exposure could represent dysbiotic vaginal microbiota and Lactobacillus crispatus exposure could represent normal vaginal microbiota. The authors revealed that transient bladder exposure to G. vaginalis-induced UPEC emergence into urine despite the UPEC not colonizing in the bladder. Moreover, the animal models that experienced G. vaginalis-induced rUTI were those that had prolonged infection after UPEC inoculation. By contrast, exposure to heat-killed G. vaginalis did not show UPEC emergence. Suggestive mechanisms how lactobacilli prevents vaginal colonization of UPEC show competitive exclusion of E. coli by strong adherence of lactobacilli to urothelial cells and decrease of vaginal pH by production of lactic acid and production of surfactants and H2O2 [34]. As a result, one of the most important events preceding rUTI is colonization of UPEC in vagina. The alterations in microbiota of vagina may result from the influence of estrogen deficiency, antimicrobial therapy, and contraceptives.
Intracellular Bacterial Community
UPEC can invade and grow rapidly within the host cells, then form biofilm-like IBCs. This process allows UPEC to survive and replicate in a safe intracellular environment, therefore avoiding host defense mechanism and antibiotics. Several researches reported that IBCs have been found in urine sediments from women and children with UTI [30,35,36].
In one study, biopsies from bladders of 33 symptomatic patients were analyzed after antibiotic therapy [37]. Sixteen of these patients (48.4%) had sterile urine cultures; however, bacteria were cultured from the others. The authors proposed that urine culture might not necessarily show the true conditions of the urothelium and that there could be a chronic niche for UPEC [30] (Fig. 1).
Type 1, P, S, and F1C pili (adhesive organelles) can promote UPEC adherence to the host cells in urothelium. The invasion process should have rearrangements of the actin cytoskeleton to envelop the adherent bacteria. After UPEC is internalized, the persistent expression of type 1 pili can develop IBCs by biofilm formation [38,39]. Intracellular biosynthesis of the auto-transporter antigen 43 might help to develop IBC by promoting UPEC aggregation [40]. Moreover, other studies using murine cystitis models have shown that entry of UPEC into urothelial cells could benefit the microbes by providing them with a niche in which to persist [7,41,42] (Figs. 1, 2).
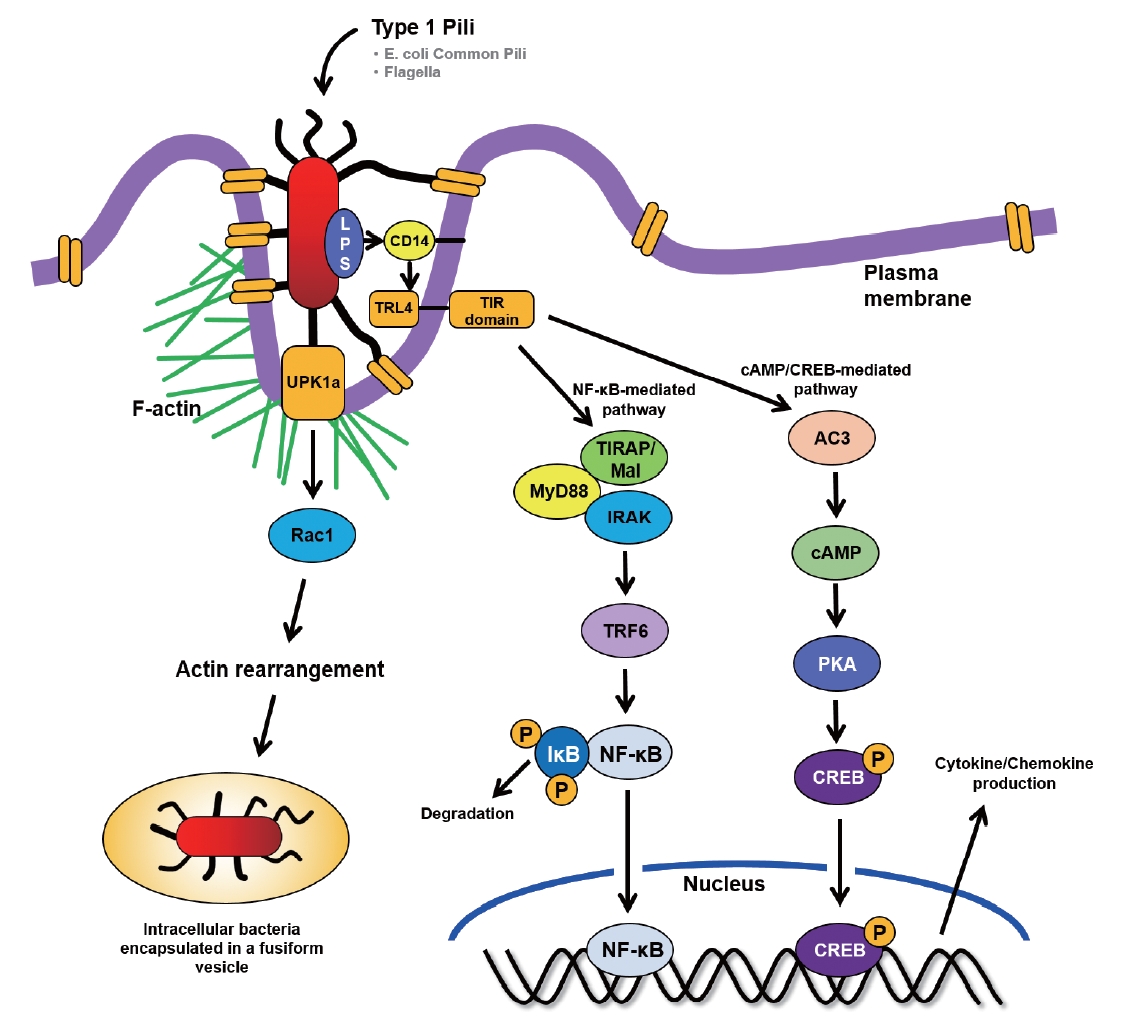
Diagrammatic illustration draws host and bacterial factors that uropathogenic Escherichia coli (UPEC) invasion and Toll-like receptor (TLR) 4-initiated signaling pathways upon ligation by surface structure of lipopolysaccharide (LPS) in bladder epithelial cells (BECs). TIR, Toll/interleukin-1 receptor; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; TIRAP, TIR adaptor protein; IRAK, interleukin-1 receptor-associated kinase; TRF6, tumor necrosis factor receptor-associated factor 6; PKA, protein kinase A; NF-κB, nuclear factor-kappa B.
Furthermore, intracellular UPEC could have ability to protected from a number of antibiotics [43,44]. In animal models, use of antibiotics can sterilize the infected urine of mice, but intracellular bacteria in the urothelium could persist for many weeks [45]. The significant effect of this process is amplification of bacterial numbers. Bacteria in the IBC can replicate as many as 105 bacteria per cell, then the bacteria in the IBC change morphology, destroy the infected cell, and infect other cells [23,38,45]. One study using urothelial cells in vitro has shown that efflux of UPEC can disrupt the host actin cytoskeleton [46]. The UPEC in cytosol of host cells enhances bacterial replication, allowing them to make inclusions with huge numbers of pathogens. The inclusions can make an association that is called bacterial factories [7,47]. Following researches mentioned the bacterial inclusions as IBCs [45,48].
Invaded UPEC also could encase in vesicles that serve as portals to move the extracellular bacteria to the cytosol [49,50]. Bishop et al. [51] reported how UPEC invaded and stayed in the intracellular space. They proposed that intracellular UPEC were contained within secretory lysosomes and the internalized UPEC were housed in vesicles enriched in Rab27b in bladder epithelial cells. After knockdown of expression of Rab27b using siRNA, UPEC invasion was significantly inhibited. Therefore, UPEC infection of the bladder epithelial cells initiated both the release of secretory lysosomes as well as the incorporation of bacteria into secretory lysosomes (Fig. 2).
Quiescent Intracellular Reservoir
On the state of infection, the host exfoliates urothelial cells into the urine. Finally, UPEC could make quiescent intracellular reservoirs (QIRs) consisted of subset of bacteria that persisted for several weeks undiscovered by the immune system [42,52]. Urothelial turnover may trigger the quiescent bacteria to replicate and lead to recurrent infection.
QIRs were reported in patients without symptoms between recurrent episodes of UTI [37]. Other studies in animal model revealed that UPEC could persist latently within urothelial cells after administration of antibiotics. They proposed that the process could induce reinfections [42,52,53]. It is suggested that the bacteria in the QIR could stay quiescently for a long time without causing UTI symptoms [7,47]. The status of the bacteria makes them difficult to find by host immune system and less susceptible to antibiotics. The revival of the QIRs can induce recurrent infections. UPECs enter a dormant state, and the quiescent condition of the UPEC offers them protected from the flow of urine, immune system surveillance, and extracellular antibiotics. QIR might exist for long time without symptoms of UTI after use of antibiotics [54]. In note, one recent study also demonstrated that dormant UPEC escaped from infected urothelium to promote recurrent UTI [33].
QIRs are importantly different from the method by which UPEC invade into the host urothelial cells during acute stage of UTI to form IBCs [45,48]. QIRs are latent infections that are able to persist for months confined within the endosomes whereas IBCs are transient, maturing over several hours within the cytosol. One significant result is the formation of antibiotic-tolerance, whereby QIRs form in the bladder within 1 week during the resolution in the acute stage of UTI. In addition, QIRs are consisted of lesser than 15%–20% bacteria existing in urothelial host cells. Decrease of bacteria titers in urine of less than 103 might show formation of QIR, in which QIRs could persist in the urothelial host cells and induce next event of infection [42,52] (Fig. 1).
Lipopolysaccharide, a Virulence Factor in UPEC
Lipopolysaccharide (LPS) may induce cytokine release in cultured urothelial cells via inflammation signaling [55]. LPS has amphipathic characteristics and it is bound to a O antigen which is a long polysaccharide chain [56]. Structure of LPS modulates life cycle of UPEC and helps to form reservoirs of UPEC, and stimulate responses of innate and adaptive immune system [57].
Umbrella cells line the bladder lumen and perform a barrier function through tight-junction proteins. The umbrella cells show specialized functions for example, the cells represent the sentinel cells in defense against UPEC through rapid release of inflammatory cytokines and mediating of impermeability in bladder to provide barrier function. Receptors of LPS in the surface of urothelium may stimulate intracellular signaling pathways. The mechanism process of activated LPS might be through a Toll-like receptor (TLR) 4-mediated increase in cytosolic calcium. This is atypical pathway to fast release mechanism of cytokines against UPEC. Interestingly, the cells shown in mouse to harbor IBCs of UPEC. The conventional antibiotics cannot eradicate LPS in IBC due to insufficient ability to penetrate urothelial cells. Due to the remnant LPS in the IBCs, symptom and pain in patients with UTI might be aggregated [58,59]. Importantly, UPEC has been implicated in inhibiting cytokine production by LPS-TLR4 signaling. Hunstad et al. [60] proposed that they isolated genes in UPEC that contributed to suppressing the cytokine responses.
UPEC has pathogen-associated molecular patterns, such as flagellin, α-hemolysin, and LPS that activate NLRP3 inflammasomes [61]. One important study showed that LPS and flagellin are able to activate inflammasomes when they existed in the urine [62]. Moreover, injection of LPS into urothelium and wall of bladder might induce symptoms of UTIs and the symptoms could be decreased by glyburide [63], clearly demonstrating that LPS can trigger a symptom of UTI. Nagamatsu et al. [64] assessed α-hemolysin expression in UPEC and reported that the upregulation of α-hemolysin induces NLRP3 and triggered pyroptosis of urothelial host cells. The process activated to exfoliate urothelial host cells which is a host defense mechanism against UPEC having α-hemolysin. However, the approach can have weak point, because exposing urothelial cells can allow the UPEC to invade deeply and make QIRs, which can lead to recurrent UTI (Fig. 2).
Multimicrobial Infection
Kline et al. [65] proposed the significance of GBS in rUTI. They showed that the bacteria might be an underestimated enemy, as they can induce complicated immune responses during acute UTI, changing UPEC-host interactions that alter the long-term outcome of UTI. GBS is a gram-positive bacteria and induced 1% of all mono-microbial UTIs, which represents about 80,000 cases annually, and can occur mainly in immunocompromised patients [66,67].
These data need additional research to evaluate GBS as a possible risk factor for UTI in immunocompromised patients, such as elderly, pregnant, and diabetic individuals. GBS might induce recurrence of infection in spite of fast clearance. Although GBS may not affect invasion or recruitment of UPEC, its existence helps with survival of them. The bacteria can promote the survival of UPEC in the early stage of UTI through increase of UPEC titers and reduction of inflammatory responses of macrophages. Taken together, the study suggested that the presence of GBS with situation of multimicrobial exposure could be a risk factor for recurrence in immunocompromised patients [65].
Mucosal Remodeling in the Bladder
Chronic bacterial cystitis in mice can trigger mucosal remodeling in the bladder that sensitizes the host immune system to recurrence. Although defense systems may prevent colonization of bacteria in the bladder, once UTI happens, induced cytokines affect the balance of immune cell defense. The immune reaction in the bladder is significant for anticipating outcome of UTI in both naïve and experienced mice. Notably, the persistence of the urothelial hurdle appears to be important to prevent recurrent UTI [68].
The protective layer of glycosaminoglycans (GAG) lining the urothelium is a part of the development of rUTIs. The GAG layer usually performs a critical role in the process of UTI. A damaged layers amplify the risk of invasion and adherence of bacteria to the urothelium [69]. Therefore, the treatment strategy of rUTI needs to include the recovery of damaged urothelium, with an intact layer that defends against invasion and adherence of bacteria. Efficacy of replacement therapy for the GAG layer against rUTI was proven in several studies. Engeler et al. [69] demonstrated that 70% of patients had no recurrence at an average follow-up of 12.4 months.
O’Brien et al. [68] demonstrated that an initial UTI is able to leave a molecular imprint on the urothelium through remodeling and transcriptional repositioning that consist cyclooxygenase-2-dependent inflammation. The outcome provides novel insight into how initial infection can cause remodeling of the innate immune system and how this can affect the outcome of recurrent infections. LPS was researched to assess the role of big-conductance potassium (BK) channel in the pathophysiology of rUTI. Specific BK inhibitors obstructed the activity of potassium conductance channel. Activity of BK channel in umbrella cells was significantly increased by LPS. The ion channel in urothelium could play a critical role in the pathophysiology of rUTI. Researchers reported that blocking the BK channel changed the mucosal and detrusor metabolomics, proposing that the BK channel in urothelium is associated with metabolism of bladder [70]. As a result, a damaged defense mechanism against microorganisms of the urothelium increases the susceptibility to subsequent bacterial infection.
WHAT DRIVES THE PATHWAYS OF RECURRENCE OR RESOLUTION OF URINARY TRACT INFECTION?
Innate and Adaptive Immunity
The urothelium reacts to infection by undergoing apoptosis and exfoliation of infected cells from the urothelial host cells. Innate and adaptive immune system are not well understood in UTI, but the defense system against bacterial challenge may explain why the majority of women who experience initial and acute UTI do not show recurrent infection [71,72]. Jaillon et al. [73] demonstrated the important role of pentraxin 3 (PTX3), a key player in the humoral and innate immunity against UTIs. PTX3-deficient animal model showed damaged response and exacerbated inflammation after UTI. Upregulation of PTX3 was induced in urothelial host cells by UPEC in a TLR4- and MyD88-dependent manner. The study also reported that increased level of PTX3 in the serum and urine of patients was associated with the severity of UTI. Moreover, they showed that polymorphisms of the PTX3 gene were correlated with susceptibility to infections. Based on these results, PTX3 could be involved in innate immunity against UTIs and have activity of ingestion of bacteria and phagosome maturation in neutrophils. In addition, PTX3-deficient neutrophils showed damaged ability of phagocytosis. The deficiency of PTX3 might show poor ability to control infection, resulting in spread of infection. Therefore, PTX3 deficiency could be associated with recurrent and chronic UTI [73].
Uroplakin IIIa Signaling
The endocytic system of UPEC invasion is very important. UPEC-mediated UPIIIa signaling has a function of initiation to induce the endocytic process in urothelial cells, it can gain access to an intracellular space [51]. Apoptosis could be a critical part of a robust innate immune system. In the process of a successful response of the immune system, the urothelial cell might perform apoptosis. Initiation of the process requires activation of UPIIIa signaling pathway. This process is associated with the recent findings that host cells in urothelium might be originated from multiple progenitors [74]. In the absence of the mechanism of clearance, invading pathogens are likely to make reservoirs such as QIR. Successful achievement of the immune-resistant environment usually requires pro-survival TLR signals that modulate the balance between apoptosis and promote cell survival. Disregarding the precise mechanisms, the balance mechanisms could affect pathophysiology of rUTI [75].
CONCLUSIONS
This review summarized the latest data in the microscopic level about the cause of UTI recurrence, such as urinary microbiota, intracelluar bacterial community, QIR, LPS, multimicrobial infection, mucosal remodeling of urothelium. By refining the way in which we consider the pathophysiology of rUTI, we are able to understand the molecular view in recurrence of UTI. Moreover, the understanding can facilitate the development of effective treatment strategy to combat the intracellular events, since the first step in development of a therapeutic strategy is understanding the exact pathophysiology of a disease.
Notes
Fund/Grant Support
This work was supported by Konkuk University Medical Center Research Grant 2020.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Conceptualization: AK, HGK
·Data curation: AK, JA, WSC, HKP
·Formal analysis: AK, JA, WSC, HKP, SK, SHP
·Funding acquisition: AK
·Methodology: AK
·Project administration: WSC, HKP, SK, SHP, HGK
·Visualization: AK, WSC, SK, SHP, HGK
·Writing-original draft: AK, JA
·Writing-review & editing: AK
Acknowledgements
I would like to express the deepest appreciation to Prof. Yeong Min Park and recurrent UTI research team.