Current Pharmacologic Strategies for Treatment of Intractable Epilepsy in Children
Article information

Abstract
Epileptic encephalopathy (EE) is a devastating pediatric disease that features medically resistant seizures, which can contribute to global developmental delays. Despite technological advancements in genetics, the neurobiological mechanisms of EEs are not fully understood, leaving few therapeutic options for affected patients. In this review, we introduce the most common EEs in pediatrics (i.e., Ohtahara syndrome, Dravet syndrome, and Lennox-Gastaut syndrome) and their molecular mechanisms that cause excitation/inhibition imbalances. We then discuss some of the essential molecules that are frequently dysregulated in EEs. Specifically, we explore voltage-gated ion channels, synaptic transmission-related proteins, and ligand-gated ion channels in association with the pathophysiology of Ohtahara syndrome, Dravet syndrome, and Lennox-Gastaut syndrome. Finally, we review currently available antiepileptic drugs used to treat seizures in patients with EEs. Since these patients often fail to achieve seizure relief even with the combination therapy, further extensive research efforts to explore the involved molecular mechanisms will be required to develop new drugs for patients with intractable epilepsy.
INTRODUCTION
Epilepsy is one of the most common neurological disorders and is characterized by recurrent, unprovoked seizures. The new epilepsy definition proposed by the International League Against Epilepsy includes patients with ≥2 unprovoked seizures separated by at least 24 hours, one unprovoked seizure with a probability of further seizure recurrence >60%, or an identified epilepsy syndrome [1]. The incidence of epilepsy shows a U-shaped pattern with 2 peaks in infancy and in elderly patients [2]. Epilepsy frequently occurs in children, especially during the first year of life [3]. Although approximately two-thirds of pediatric patients with epilepsy can be successfully controlled with appropriate antiepileptic drugs (AEDs) and achieve sustained seizure freedom, the remaining one-third suffers from uncontrolled epilepsy even with a combination of AEDs; these patients are diagnosed with refractory epilepsy [4]. Given that brain development is actively ongoing in childhood, refractory epilepsy in children is a serious problem that severely affects the patients’ quality of life. Moreover, refractory epilepsy frequently contributes to high mortality and a wide variety of morbidities, including impaired intellectual, behavioral, and social outcomes as well as developmental stagnation or regression [5], which require intensive investigation. Therefore, in this review, we introduce the 3 most common intractable epilepsy syndromes in children: Ohtahara syndrome (OS) during the neonatal period, Dravet syndrome (DS) in infancy, and Lennox- Gastaut syndrome (LGS) in childhood and adolescence. In addition, we cover current knowledge on the molecular pathophysiology and treatment strategies for epileptic encephalopathies (EEs) in childhood.
COMMON CHILDHOOD EPILEPTIC ENCEPHALOPATHIES
Ohtahara Syndrome
OS, which is also known as early infantile epileptic encephalopathy with burst-suppression, is the earliest developing age-dependent EE diagnosed in children (Table 1). Seizures appear during the first 3 months of life, frequently within 10 days of birth [6]. The seizure phenotype includes mostly tonic spasms with or without clustering regardless of the type of sleep cycle. The electroencephalogram (EEG) pattern is characterized by a periodic burst-suppression pattern during both waking and sleeping states, which is critical for diagnosis of OS. Most seizures in OS patients are intractable to conventional AEDs, which contributes to the very poor prognosis seen in this disorder. Half of OS patients die within the first year of life, while the survivors commonly develop profound psychomotor impairments and significant intellectual disabilities [7]. As the child ages, OS often transforms into West Syndrome, while a far fewer number evolves into LGS [6,7]. The underlying etiology of OS patients is very heterogeneous, ranging from genetic defects, mitochondrial abnormalities, and metabolic disability to structural abnormalities of the brain [6]. However, the neurobiological backgrounds of OS patients are largely unknown, as only a few studies have shown a possible link between genetic mutations, such as voltage-gated sodium channel α2 (SCN2A), voltage-gated potassium channel subfamily Q member 2 (KCNQ2), Aristaless-related homeobox, syntaxin-binding protein 1 (STXBP1), and cyclin-dependent kinase-like 5, and OS pathophysiology [8,9].

Summary of the most common epileptic encephalopathies in pediatrics
Dravet Syndrome
DS is a well-known, medically intractable type of epilepsy that is characterized by frequent episodes of prolonged seizures (Table 1). DS usually presents in the first year of life with generalized clonic or hemiclonic seizures triggered by fever [10]. Gradually, multifarious seizure types (including myoclonic, absence, focal, generalized tonic-clonic, and atonic drop seizures) can appear between the ages of 1 and 4 years [11]. Although development is normal in the first year of life, intellectual, behavioral, and motor disabilities become apparent from the second year of life [11].
About 75%–80% of patients diagnosed with DS have a de novo mutation within the gene for sodium channel α1 subunit (SCN1A), a voltage-gated sodium channel. The location and the features of the mutation in this SCN1A gene are quite varied and can include a nonsense mutation, a missense mutation, a frameshift mutation, a splice‐site mutation, and gross chromosomal rearrangements [12,13]. Several other genes, such as the α1 subunit and the γ2 subunit of the γ-aminobutyric acid (GABA) type A receptor (GABRA1, GABARG2), hyperpolarization‐ activated cyclic nucleotide‐gated channel 1, voltage‐gated potassium channel subfamily A member 2 (KCNA2), chromodomain helicase DNA-binding protein 2 (CHD2), STXBP1, SCN2A, voltage-gated sodium channel α8 and β1 subunits (SCN8A and SCN1B), and protocadherin-19, have been reported in DS patients where SCN1A mutations were not found, which suggests that complex molecular mechanisms underlie DS pathophysiology [9,14,15].
Lennox-Gastaut Syndrome
LGS is a rare but severe EE that accounts for 1%–10% of all cases in children [16,17]. LGS patients start to show seizures usually between 1 and 7 years of age, which progressively aggravates their normal development (Table 1). Phenotypically, multiple seizure types are often seen, including atypical absence, atonic or drop attacks, tonic, myoclonic, and generalized tonicclonic seizures. During these seizures, bursts of slow spike waves or generalized paroxysmal fast activities can be detected on EEG [16]. LGS is defined by the triad of multiple drug-resistant seizure types, characteristic abnormal EEG findings, and intellectual disability [16].
The etiology of LGS can be either identifiable (genetic-structural- metabolic) or unknown [17]. Identifiable etiologies account for 65%–75% of cases and are derived mainly from diffuse cerebral injuries, including tuberous sclerosis complex, infections (such as meningitis and encephalitis), birth injury/trauma, developmental brain malformations, and hereditary metabolic disorders. The remaining 25% of patients can be classified with an unknown (cryptogenic) type of LGS. Although the molecular mechanisms of LGS are largely unknown, several genetic studies have found de novo mutations in certain genes, including SCN1A, SCN2A, SCN8A, glutamate ionotropic receptor N-methyl- D-aspartate (NMDA) type subunit 2B (GRIN2B), STXBP1, CHD2, forkhead box G1, dynamin 1, and the β3 subunit of the GABA type A receptor (GABRB3) [9].
MOLECULAR MECHANISMS OF INTRACTABLE PEDIATRIC EPILEPSIES
Our current understanding of the molecular mechanisms of EEs in pediatrics remains in its infancy. Although the advent of molecular genetics has shed light on clues about crucial players responsible for the excitation/inhibition (E/I) balance in the brain, there is a lack of consistent experimental models that recapitulate all the critical clinical features. In this review, we briefly explain the basic molecular mechanisms of the E/I imbalance that causes seizures in association with key molecules known to be deregulated in intractable epilepsies in children (Table 1).
The E/I Imbalance
The pathophysiological mechanisms that lead to seizures are grounded in hyperexcitability resulting from an E/I imbalance at single-cell and at network levels. Excessive excitation, reduced inhibition, or both can attribute to a hyperexcitable and hypersynchronous state of the brain that can increase the propensity for seizures [18].
At the cellular level, the action potential is propagated by changing the currents of voltage-gated ion channels in both excitatory and inhibitory neurons. In particular, opening of voltage-gated sodium channels can lead to membrane depolarization, followed by rapid inactivation. During membrane depolarization, potassium channels open, resulting in membrane hyperpolarization that requires more time to generate new action potentials. Sodium channels then recover from their inactivated state as the membrane potential is fully repolarized. Under physiological conditions, the refractory period derived from sodium channel inactivation to potassium current-induced membrane hyperpolarization can cause neurons to be unaffected by depolarizing signals. Therefore, any changes to promote membrane depolarization or to reduce the refractory period can lead to excessive excitation of neurons.
An individual neuron with a robust action potential can activate adjacent neurons, causing numerous neurons to discharge simultaneously [19]. Activation of voltage-gated calcium channels promotes the release of neurotransmitters as well as neuronal depolarization. Once neurotransmitters (including glutamate and GABA) are released, various postsynaptic ligand-gated ion channels can be activated, modulating signal transmission. For example, NMDA and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors contribute to generate postsynaptic excitatory potentials and thereby recruit additional excitatory neurons to be activated [20]. In contrast, GABA receptors can prevent neuronal hyperexcitability by increasing chloride currents that help to escalate the action potential threshold of postsynaptic neurons, mediating any surrounding inhibition [21]. Thus, either activation of glutamate receptors or inhibition of GABAergic neurotransmission at the network level can lead to a hyper-synchronization state of the brain, which is prone to seizure generation.
Potential Molecular Mechanisms Associated With Intractable Epilepsies in Children
In this part of the review, we introduce essential voltage-gated ion channels, synaptic transmission-related proteins, and ligand-gated ion channels that are dysregulated in OS, DS, and LGS and are thought to contribute to the accompanying E/I imbalance in the brain (Fig. 1).
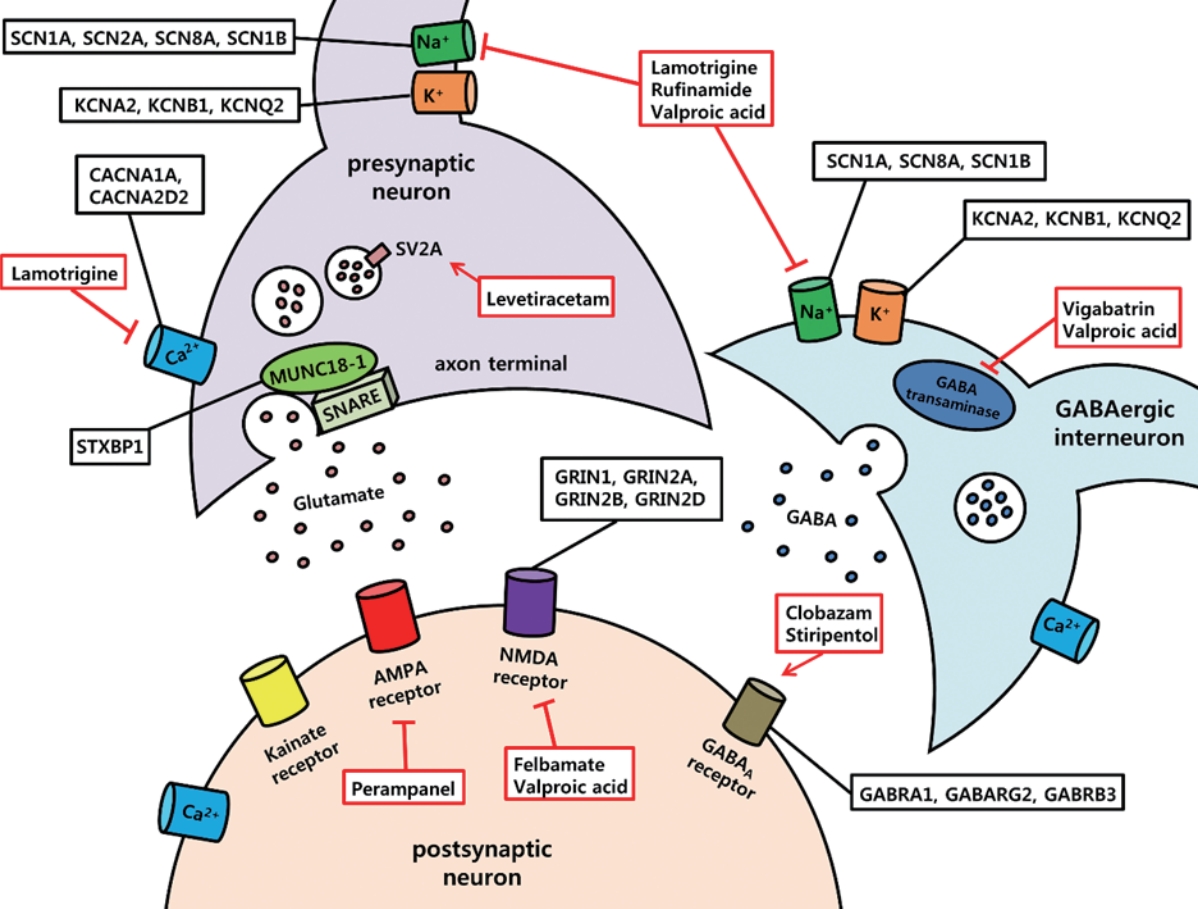
Essential molecular targets and antiepileptic drugs in intractable pediatric epilepsies. Voltage-gated ion channels (sodium, potassium, and calcium), synaptic transmission-related proteins, and ligand-gated ion channels (glutamate receptors, γ-aminobutyric acid type A receptor) are frequently mutated in epileptic encephalopathies. Examples of mutated genes in Ohtahara syndrome, Dravet syndrome, and Lennox-Gastaut syndrome are described in black box. In addition, crucial antiepileptic drugs that are currently available for epileptic encephalopathies are illustrated in red box. Red arrow indicates potentiating effects by drugs, and red lines with blunt ends indicate inhibitory action of drugs. SCN1A, voltage-gated sodium channel α1 subunit; SCN2A, voltage-gated sodium channel α2 subunit; SCN8A, voltage-gated sodium channel α8 subunit; SCN1B, voltage-gated sodium channel β1 subunit; KCNA2, voltage‐gated potassium channel subfamily A member 2; KCNB1, voltage‐gated potassium channel subfamily B member 1; KCNQ2, voltage-gated potassium channel subfamily Q member 2; CACNA1A, voltage-gated calcium channel subunit alpha 1A; CACNA2D2, voltage-gated calcium channel auxillary subunit alpha 2 delta 2; SV2A, synaptic vesicle glycoprotein 2A; SNARE, soluble N-ethylmaleimide sensitive factor attachment protein receptor; STXBP1, syntaxin-binding protein 1; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-D-aspartate; GABA, γ-aminobutyric acid; GRIN1, glutamate ionotropic receptor N-methyl-D-aspartate type subunit 1; GRIN 2A, glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2A; GRIN2B, glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2B; GRIN2D, glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2D; GABRA1, γ-aminobutyric acid type A receptor α1 subunit; GABARG2, γ-aminobutyric acid type A receptor γ2 subunit; GABRB3, γ-aminobutyric acid type A receptor β3 subunit.
Voltage-Gated Sodium Channels
Voltage-gated sodium channels are composed of an α subunit encoded by 9 genes (SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN8A, SCN9A, SCN10A, and SCN11) and one or more β subunits that are encoded by 4 genes (SCN1B–SCN4B) [22]. Mutations in SCN1A, SCN2A, SCN8A, and SCN1B have been reported in EEs [9]. SCN1A mutation (encoding Nav1.1) is one of the most crucial causative genes in DS and LGS [9]. Nav1.1 is expressed widely both in excitatory neurons and in interneurons located in the neocortex, hippocampus, and cerebellum [23]. Interestingly, previous studies that have used SCN1A knockout mice and induced pluripotent stem cell-derived neurons from DS patients have described that loss-of-function (LOF) of Nav1.1 (especially in GABAergic-inhibitory neurons) leads to hyperexcitability of excitatory neurons due to deficits in action potential firing of the interneurons, which is known as the “interneuron hypothesis” [24-26]. However, another study that used DS patient-derived neurons reported an increased sodium current in multiple cell types including both GABAergic interneurons and glutamatergic neurons based on morphology [27]. Thus, further studies are required for a clear understanding of DS pathophysiology. With regard to the β subunit of voltage-gated sodium channels, SCN1B mutations have been reported mainly in patients with DS [28,29]. SCN1B-based mouse models of DS demonstrated a heightened susceptibility to seizures [29] with the increased action potential firing in excitatory pyramidal neurons but no changes in interneurons [30]. Considering all these seemingly discordant reports, it can be inferred that seizures arise from multiple functional defects of voltage-gated sodium channels that can trigger an E/I imbalance.
Voltage-Gated Potassium Channels
Voltage-gated potassium channels are composed of 4 α subunits that form the channel pore and auxiliary β subunits [31]. There are 12 subfamilies (Kv1-12) of voltage-gated potassium channels that display approximately 40 α subunits [31]. Mutations in several potassium channel genes, including KCNA2 (Kv1.2), KCNB1 (Kv2.1), and KCNQ2 (Kv7.2), have been associated with EEs [32,33]. Kv2.1 is expressed in both pyramidal neurons and interneurons and mediates delayed rectifier potassium current [34,35]. By inducing membrane repolarization, Kv2.1 can suppress high-frequency neuronal firing [36]. Although a previous study that used Kv2.1 knockout mice showed no spontaneous seizures, Kv2.1 deletion exhibited hypersensitivity to chemo-convulsant injections, in addition to accelerating seizure progression [35]. Other genes, such as KCNQ2 or KCNQ3, can cause either a homogeneous (Kv7.2) or a heterogeneous (Kv7.2/Kv7.3) complex to become co-localized at the axon’s initial segment [37]. Kv7.2 or Kv7.2/Kv7.3 complexes can form low-threshold voltage-gated K+ channels or M-channels and regulate the neuronal membrane potential via slow, noninactivating muscarinic currents [38]. Thus, open state of Kv7.2 or Kv7.2/Kv7.3 complexes can inhibit initiation of the action potential and suppress repetitive neuronal firing [38,39]. This notion was supported by a mouse line that showed spontaneous mutations involving KCNQ2, demonstrating increased neuronal excitability [40]. Therefore, deficits in membrane repolarization through mutated voltage-gated potassium channels might cause a failure of natural antiepileptic mechanisms of the brain and lead to an E/I imbalance.
Voltage-Gated Calcium Channels
Voltage-gated calcium channels are classified into 2 major groups based on threshold of membrane action potential: high voltage-activated (HVA) calcium channels (L-, N-, P-, Q-, R-types) and low voltage-activated calcium channels (T-type) [41]. Each voltage-gated calcium channel consists of an α1 subunit that forms the pore, a complex of α2 and δ subunits, an intracellular β subunit, and (in some cases) a γ subunit [41]. While L-type calcium currents play major roles in excitation of postsynaptic neurons, N-, P/Q-, and R-type calcium currents are involved in rapid synaptic neurotransmitter release [42]. In LGS and OS, mutations in calcium voltage-gated channel subunit alpha 1A (CACNA1A) and calcium voltage-gated channel auxiliary subunit alpha 2 delta 2 (CACNA2D2) have been reported [9]. In particular, CACNA1A encodes the α1 subunit of the P/Q type calcium channel, which is important for neurotransmitter release. As expected, CACNA1A LOF mutations in cortical interneurons demonstrated reduced GABA release, supporting the severe epileptic phenotype in mutant mice [43]. Thus, impaired ability of CACNA1A might contribute to cellular excitability and produce seizures resulting from increased neuronal firing by altering inhibitory neurotransmission.
Syntaxin-Binding Protein 1
A continuous cycle of synaptic vesicle formation, presynaptic neurotransmitter release, synaptic vesicle recycling, and activation of postsynaptic receptors is essential for synaptic transmission [44]. Multiple molecular players (such as dynamin, synaptic vesicle glycoprotein 2A (SV2A), synapsin, and syntaxin) are involved in this multistep process [45-47]. The STXBP1 gene encodes the presynaptic protein MUNC18-1, which is a trafficking protein that regulates synaptic vesicle docking and fusion through interactions with the soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) complex and plays a critical role in neurotransmitter secretion [48-51]. STXBP1 mutations have been shown to cause EEs, including DS, LGS, and OS [9]. Decreased expression of STXBP1 can reduce the synaptic vesicle pool size, which is more common in interneurons than in glutamatergic neurons [49,51], suggesting a net hyperexcitability leading to seizures.
Glutamate Receptors
Glutamate receptors are grouped into 2 families: (1) ligand-gated ion channels that produce excitatory postsynaptic currents and (2) metabotropic receptors that are G protein-coupled receptors [52]. Three subtypes of ionotropic glutamate receptors are further classified: AMPA receptors (GluA1–GluA4), kainate receptors (GluK1–GluK5), and NMDA receptors (GluN1, Glu- N2A-GluN2D, GluN3A, and GluN3B). Among these, epilepsyrelated mutations in glutamate receptors include GRIN1, GRIN2A, GRIN2B, and GRIN2D, which encode the GluN1, Glu- N2A, GluN2B, and GluN2D subunits, respectively. Specifically, GRIN2B mutations have been identified in patients with LGS and West Syndrome [9]. However, little is known about the neurobiological impact of these mutations in EEs, which warrants further investigation.
GABA Type A (GABAA) Receptors
GABAA receptors are ligand-gated ionotropic receptors that can mediate phasic (synaptic) or tonic (extrasynaptic) inhibitory neurotransmissions by allowing inward chloride flow causing postsynaptic hyperpolarization [53]. Structurally, heteropentameric GABAA receptors are composed of various combinations of subunits (α1-6, β1-3, γ1-3, δ, ε, θ, and ρ1-3); the most abundant subtype contains 2 α-, 2 β-, and 1 γ-subunit [53]. In DS, OS, and LGS, de novo mutations of the GABAA receptor genes GABRA1, GABRG2, and GABRB3 that encode the α1, γ2, and β3 subunits, respectively, have been reported [9,14,15,54,55]. Impaired GABAergic inhibition by GABRA1 mutations could reduce the total surface expression of GABAA receptors in addition to decreasing the effectiveness of neurotransmitters [56]. Compared to GABRA1, GABRG2 plays several complicated roles, given that heterozygous GABRG2 knockout mice demonstrated an absence of seizures, while heterozygous GABRG2 knock-in mice showed more severe epilepsy phenotypes [57,58]. Finally, the GABRB3 mutations present in patients with LGS reduced GABA-evoked currents by disrupting the GABA binding site but led to no changes in the surface expression of GABAA receptors [55]. Similarly, GABRB3 knock-in mice showed reduced inhibitory postsynaptic currents with no alteration in GABAA receptor trafficking [59]. Collectively, disruption of GABAA receptor subunits can impair network-level surrounding inhibition and cause susceptibility to seizures.
PHARMACOLOGICAL APPROACHES TO INTRACTABLE EPILEPSIES IN PEDIATRIC PATIENTS
Most patients with EEs (including OS, DS, and LGS) are highly refractory to AEDs, so a combination therapy of more than 2 AEDs is recommended. Therapeutic strategies are aimed to reduce the frequency of seizures to improve patient quality of life and to minimize the side effects of AEDs. Many currently available AEDs are designed to restore the E/I balance in the brain and include drugs that can modulate voltage-gated ion channels, those that can modulate synaptic neurotransmission, drugs that can inhibit glutamate-mediated excitation, substances that can enhance GABA-mediated inhibition, and drugs that have multiple mechanisms (Fig. 1, Table 1).
Drugs That Modulate Voltage-Gated Ion Channels
Lamotrigine, which is typically combined with valproic acid, has been used for treatment of LGS [60,61]. Although the mechanism of lamotrigine is not fully understood, it is thought to stabilize the neuronal membrane by protracting the inactivated state of sodium channels [60]. Another proposed mechanism of lamotrigine is related to inhibition of the HVA calcium channel [62]. These channels can mediate rapid neurotransmitter release by controlling the influx of calcium into the presynaptic terminals [42]. Thus, reduction in HVA calcium currents by lamotrigine can decrease the release of neurotransmitters.
Rufinamide is used as an adjunctive treatment of the seizures that are associated with LGS [63]. This drug also acts on voltage-gated sodium channels by slowing their recovery from the inactive state, thereby limiting the excessive firing of sodiumdependent action potentials in neurons [64,65]. Rufinamide also has an additional mechanism that can inhibit a subset of glutamate receptors (such as mGluR5) at higher concentrations, supporting its antiseizure effects [66].
Drugs That Modulate Presynaptic Neurotransmitter Release
Levetiracetam can be used as an adjunctive treatment for various EEs [67-70]. Its mechanism is related with SV2A, which is mainly located in the presynaptic terminals and promotes synaptic vesicle release and recycling [71,72]. Specifically, levetiracetam can enter the recycling synaptic vesicles and decrease the readily releasable pool of vesicles by inhibiting SV2A and eventually reducing excitatory postsynaptic currents [73]. Interestingly, levetiracetam can decrease activity-dependent neurotransmitter release while sparing the normal synaptic transmission, which reduces the number of potential adverse effects [74].
Drugs That Inhibit Glutamate-Mediated Excitation
Perampanel is a noncompetitive AMPA receptor antagonist [75] with demonstrated efficacy in refractory epilepsies, including LGS [76,77]. Although definite mechanisms of the antiepileptic effects of perampanel have not been established, the drug is known to promote a closed state of the AMPA receptor, preventing opening of the ion channel [78] and resulting in inhibition of neuronal excitation.
Felbamate has a particular demonstrated efficacy on LGS, which selectively blocks NMDA receptors (including the NR2B subunit) and leads to reduction in glutamatergic transmission [79]. Since the expression of the NR2B subunit is not as widespread as that of other subunits throughout the brain, felbamate can reduce the potential side effects derived from inhibition of NMDA receptors. Felbamate can potentiate GABAergic inhibition through allosteric interaction with GABAA receptors [79, 80]. In addition, by blocking the sodium channel, felbamate can reduce sustained repetitive firing [81], which suggests that this drug has multiple mechanisms of action [82].
Drugs That Enhance GABA-Mediated Inhibition
Drugs that enhance GABAergic inhibition mostly target GABAA receptors, GABA metabolic enzymes, or GABA transporters [83]. Clobazam is used frequently as the first-line treatment of DS along with valproic acid or as an adjunctive treatment in LGS [84-86]. Clobazam is a 1,5-benzodiazepine that acts mainly through activation of postsynaptic GABAA receptors, inducing phasic inhibition [87].
Stiripentol can be added as a second‐line drug for DS [85] and is an aromatic allylic alcohol. Its main mechanism of action is induction of inward chloride currents that are created by increasing the open duration of GABAA receptors [88]. This substance can also increase synaptic GABA level through interference in GABA reuptake and metabolism [89].
Vigabatrin showed favorable responses in OS and West Syndrome [90,91]. This drug is an irreversible inhibitor of GABA transaminase that metabolizes GABA into succinic semialdehyde, resulting in the increase of synaptic GABA concentrations [92,93]. Vigabatrin can also enhance tonic inhibition via GABA transporter reversal due to high levels of intracellular GABA [94].
Broad-Spectrum Drugs
Valproic acid is a pleiotropic drug with multiple mechanisms of action, including inhibition of voltage-gated sodium and T-type calcium channels, potentiation of GABAergic inhibition, and inhibition of NMDA receptor-mediated neuronal excitation [95]. Due to its wide mechanisms of action, valproic acid often is used as an initial treatment in DS and as an adjunctive treatment for LGS and OS [84,85,96,97].
CONCLUSIONS
Despite recent technological advancements in next-generation sequencing that have identified many new mutations in intractable pediatric EEs, the neurobiological mechanisms linking these mutations and seizure generation are not yet fully appreciated. In this review, we described the clinical features of the 3 most common EEs and their basic molecular mechanisms with an emphasis on several essential molecules that are dysregulated in OS, DS, and LGS. In addition, we explored the pharmacological options currently available for treating intractable EEs in childhood. Since no effective drug has demonstrated superior efficacy for seizure control in EEs (including OS, DS, and LGS), more extensive research efforts are required to cure these devastating diseases.
Notes
Fund/Grant Support
This study was supported by the National Research Foundation of Korea (NRF) (grant nos. 2019R1A2C1003958, 2019K2A9A2A08000167 to KOC).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
- Conceptualization: KOC
- Data curation: JUM
- Formal analysis: JUM
- Funding acquisition: KOC
- Methodology: JUM
- Project administration: KOC
- Visualization: JUM, KOC
- Writing-original draft: JUM, KOC
- Writing-review & editing: KOC