INTRODUCTION
Medical dogma holds that the urine of healthy asymptomatic individuals is sterile; however, bacteria have been cultured in the absence of symptoms. Therefore, the presence of bacteria in the urine is not sufficient to cause urinary tract infection (UTI). Conversely, some patients present with symptoms suggestive of UTI who do not have positive urine cultures. The apparent incongruence between the presence of bacteria and human health and disease is becoming better understood through the application of highly sensitive genomic analytical techniques. Next-generation sequencing (NGS), for instance, has identified a diverse ecosystem, termed ‘the human microbiome,’ which includes the prokaryotes, viruses and microbial eukaryotes that populate the human body, and which fluctuates depending on the clinical scenario. As genomic tools become commonplace, it is likely that the information gleaned from them should refine our long-established perspectives on human health.
Asymptomatic bacteriuria (ASB) is a recognized clinical entity defined by growth of a single uropathogen at >105 colony-forming unit (CFU)/mL in 2 consecutive midstream voided urine samples from females and one midstream voided urine sample from males [1]. The prevalence of ASB varies by age, sex, and comorbidities (Table 1). ASB is present in 3% of premature infants, 1% of full-term infants, 0.8% and 0.3% (respectively) of preschool-aged girls and boys, and up to 5% of schoolage girls. By 15 years, 30% of young women and up to 100% in catheterized patients have ASB [2-6]. The most common organism isolated in samples from patients with ASB and normal urinary tracts is Escherichia coli [7]. Among patients with comorbidities, non-E. coli organisms within Enterobacteriaceae and gram-positives are found [7]. Pseudomonas aeruginosa and Proteus are routinely cultured from institutionalized patients and those with catheters [7]. It can be postulated that the specific host-pathogen interactions necessary to cause symptoms are not present in these circumstances and instead a commensal relationship develops, much like exists in the gastrointestinal tract. Complicating the discourse on ASB is the observation that as many as 30% of patients initially diagnosed with ASB either are symptomatic having not fully reported symptoms or become symptomatic during follow-up [8,9]. Conversely, individuals may present urinary complaints suggestive of UTI who have negative urine cultures by standard aerobic cultivation techniques. Enhanced quantitative urine culture has identified microbes in up to 90% of standard cultures deemed “no growth” [10,11]. Treatment of cryptic microbes not identified by urine culture has resulted in clinical improvement. Thus, standard urine culture can mislead the clinician into overtreating or undertreating patients based on the traditional paradigm.
We propose that the clinical protocol for the evaluation of UTI which is based on standard urine culture deserves refinement and that the initial step should be to understand the nature of the microbiome of the urinary tract in asymptomatic individuals beyond the limited information derived from traditional cultivation techniques. We hypothesize that NGS of 16S ribosomal RNA (rRNA) amplicons of asymptomatic adult urine specimens can help to define the ‘healthy’ urine microbiome and can be used as a reference standard against which future comparisons could be made. Seeking to expand upon earlier work, we have investigated here whether there are any regional differences in the microbiota of the urethra versus the bladder and sought to understand those differences by obtaining paired samples (voided and catheterized) from each of 20 asymptomatic adults.
MATERIALS AND METHODS
Patients
Participants were recruited from the MedStar Georgetown University Hospital via flyers and announcement boards as part of this Institutional Review Board approved study (Georgetown Hospital/MedStar, IRB 2012-1416). To be included in the study, the individual had to have no known genitourinary pathology, to not be suspected of having a UTI, and to be at least 18 years of age. All subjects were queried as to the presence or absence of urinary symptoms at the time of urine collection. The study clinician confirmed the determination that “no UTI” was present. Consented subjects provided a brief medical history by using a structured questionnaire that included demographic information, genitourinary history (previous surgeries, genitourinary conditions, and current medications), as well as relevant family history. Twenty asymptomatic adults participated, comprised of 6 females (mean age, 21.2 years; range, 24–40 years) and 14 males (mean age, 31.7 years; range, 20–61 years.). Among the females, the racial distribution was Caucasian (3), African American (2), and Asian (1). Among the males, the racial distribution was White (6), African American (4), Asian (2), and Hispanic (2).
Urine Acquisition and Preparation
The 20 healthy subjects with normal bladder function described above-provided urine samples for (1) urinalysis and urine culture, and (2) 16S rRNA amplicon sequencing. Urine samples were collected by midstream voided collection first, followed by collection by sterile catheterization of the bladder after the subjects had the opportunity to ingest water and allow the bladder to refill naturally. Collected urine was immediately placed at 4°C and processed within 6 hours for standard urinalysis and urine culture in the clinical diagnostics laboratory as well as for preparation for sequencing. Urine samples were processed for sequencing by centrifugation at 4°C, 5,000 ×g, for 20 minutes. The pellet was recovered, retaining –1 mL of residual urine supernatant to avoid disturbing the urinary pellet. Addition of –10 mL ice-cold phosphate-buffered saline was followed by gentle shaking of the tube and further centrifugation at 4°C, 5,000 ×g, for 15 minutes, which was repeated once. The wet urinary pellet was frozen at -80°C until lysis was performed using 2 mL of 10mM ammonium bicarbonate buffer containing 0.1% Triton-X100, 0.5% octylglucoside, 5-µg/mL leupeptin, 10mM ethylenediaminetetraacetic acid and 2mM 2,6-Dichlorobenzamide. The suspensions were heated to 85°C for 5 minutes and sonicated at amplitude 4 (Misonex 3000 sonicator) in 30 seconds on/15 seconds off cycles 10 times on ice. The suspension was centrifuged at 4°C, 16,100 ×g, for 15 minutes and the supernatant recovered.
Urinalysis
Urine samples were assessed at Quest Diagnostics Nichols Institute (Chantilly, VA, USA) using standard clinical microbiology semiquantitative chemical testing using commercial disposable test strips for glucose, bilirubin, ketone, specific gravity, blood, pH, protein, urobilinogen, nitrite, and leukocyte esterase. After centrifuging for 5 minutes, microscopic examination for and quantification of white blood cells, red blood cells, epithelial cells, yeast, bacteria, Trichomonas vaginalis, sperm cells, mucous filaments, and crystals were performed using standard techniques.
Urine Culture
Standard urine culture microbiology was performed. Each sample was mixed thoroughly and the top of the container removed. A calibrated wire-inoculating loop (calibrated to deliver 0.01 mL per loopful) was flamed and allowed to cool without coming in contact with any surface. The sterile loop was inserted into the urine sample vertically and urine was allowed to adhere to the loop. The loopful of urine was inoculated onto MacConkey agar plates using standard methods. Similarly, a second loopful was collected and inoculated onto a blood agar plate. The plates were incubated aerobically at 35°C–37°C for at least 24 hours. The colonies and CFUs were counted by multiplying by 100 (since a 0.01-mL loop was used).
Sample Preparation and DNA Isolation
Depending on the size of the pelleted material, genomic DNA was isolated either with the DNeasy Kit (Qiagen, Venlo, the Netherlands) using manufacturer’s protocols for gram-negative bacteria or with the QIAmp DNA Micro Kit (Qiagen) using manufacturer’s protocols for DNA isolation from urine. Purified DNA was quantified using NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Fractions of human and bacterial DNA in each sample were determined using Femto Human and Femto Bacterial DNA quantification kits (Zymo Research, Irvine, CA, USA) according to manufacturer’s instructions.
16S rRNA Gene Amplification and Sequencing
V4 regions of 16S rRNA genes were amplified using primers 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGCCAGCMGCCGCGGTAA-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTWTCTAAT-3′ and the following reagent concentrations: 20mM TrisHCl (pH, 8.4), 50mM KCl, 1.5mM MgCl2, 200µM of each dNTP, 2µM of each primer, 1% glycerol, 0.3-U AccuPrime Taq polymerase (Thermo Fisher Scientific) and 25 ng of template DNA in 20 µL total volume. Amplification conditions were 2 minutes at 95°C initial denaturation followed by 30 cycles of 20 seconds. denaturation at 95°C, 15 seconds annealing at 55°C and 5 minutes extension at 72°C, and a 5-minute final extension at 72°C. Amplification products are purified with the AMPure XP system (Beckman Coulter, Brea, CA, USA) and their size was verified with the DNA 1,000 Kit (Agilent Technologies, Inc., Santa Clara, CA, USA). Indexing and pooling of amplification products were carried out according to Illumina’s 16S Metagenomic Sequencing Library Preparation protocol. Resulting library was sequenced using Illumina MiSeq Reagent Kit v2 (500 cycles).
Statistical Analysis
Raw FASTQ files were processed in mothur v1.35.1 [12]. Default settings were used to minimize sequencing errors [13]. Clean sequences were aligned to the SILVA123-based bacterial reference alignment at http://www.mothur.org. Chimeras were removed using uchime [14] and nonchimeric sequences were classified using a naïve Bayesian classifier [15]. Sequences were clustered into Operational Taxonomic Units (OTUs) at the 0.03 threshold (species level). OTU sequence representatives and taxonomy were imported (BIOM format) into Quantitative Insights Into Microbial Ecology (QIIME) [16] for subsequent analyses. Samples were subsampled (rarefaction analysis) to the smallest sample size (11,479 reads/sample) to remove the effect of sample size bias on community composition. Trees for phylogenetic diversity (PD) calculations were constructed using FastTree and midpoint rooting [17]. Alpha-diversity was estimated using the abundance-based coverage estimator (ACE), Shannon, and Faith’s PD indices [18]. Phylogenetic beta-diversity Unifrac metrics (unweighted and weighted Unifrac) were calculated between pairs of samples. Dissimilarity between samples was explored using principal coordinates analysis (PCoA) and both Unifrac distances. Linear mixed-effects (LME) models analysis was applied to both alpha-diversity indices and genera proportions (response) while accounting for nonindependence of subjects (random effect), predictors (method of urine collection and sex), and one covariable (age). Beta-diversity Unifrac indices were compared using permutational multivariate analysis of variance (adonis) as implemented in the vegan R package [19] and the same predictors and covariable above. Voided and catheterized sample pairs were also compared for each of the 20 participants using the Fisher exact test. Significance was determined through 10,000 permutations. Bonferroni or Benjamini-Hochberg false discovery rate multiple test correction methods were applied. All analyses were performed in mothur, QIIME, and RStudio [20].
RESULTS
Urinalysis and Urine Culture Findings
Among the 20 participants, pyuria >5 white blood cells per high power field was seen in only 1 male (catheterized sample only) and 1 female (voided sample only) (Table 2). In the male with pyuria, leukocyte esterase determination was “trace,” while in the female, it was “2+.” Only 5 participants demonstrated bacterial growth by standard cultivation, 4 females and 1 male (Table 3).
Sequencing
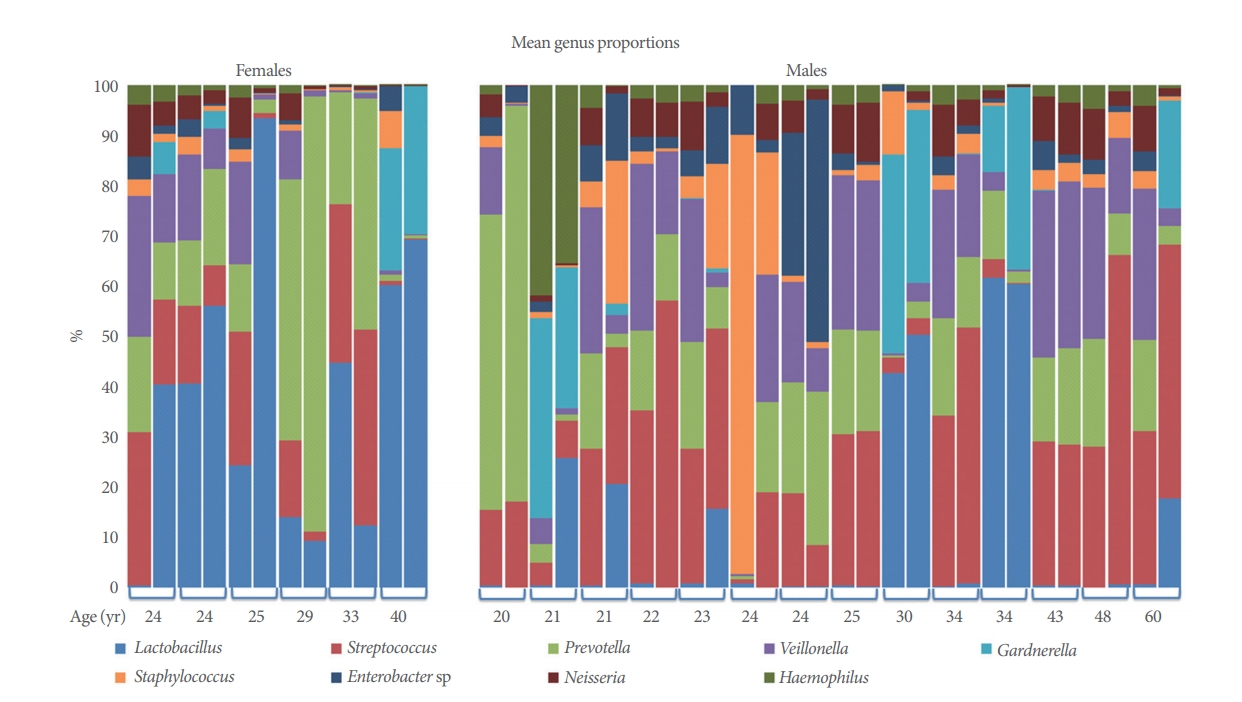
A total of 4,154,214 sequences ranging from 11,479 to 462,312 sequences per sample (mean, 103,855.4; median, 78,199.5) were obtained after quality control analyses and OTU filtering. From these data, we identified a total of 1,612 OTUs. All organisms identified by standard urine culture were also identified by 16S amplification; however, NGS also detected bacteria not identified by standard cultivation. The 9 most predominant genera across all samples are detailed (Table 4, Fig. 1). Lactobacillus and Streptococcus were the most abundant species (≥14% of all reads).
Voided (“urethra”) versus catheterized (“bladder”) urine samples: Microbial mean proportions (abundances) of all the 9 predominant (>2.5%) bacterial genera varied between urethra and bladder (Table 4). Urethral samples showed the highest abundance of Veillonella, Staphylococcus, and Neisseria, while bladder samples showed the highest abundance of Lactobacillus, Streptococcus, and Gardnerella. The other 3 genera varied less between groups. Our LME analyses showed significant differences in abundance for Veillonella (P =0.008), Neisseria (P=0.022), and Lactobacillus (P=0.041). Voided and catheterized microbiomes share all the dominant ( >1%) genera and OTUs but in similar or different proportions. Hence, urethra and bladder microbiomes do not differ in taxonomic composition, but rather in taxonomic structure.
Alpha-diversity (intra-sample) indices (Shannon, ACE, and PD) varied across urine phenotypes (Fig. 2), but only Shannon estimates, which showed less diversity for bladder, were significantly different (P=0.017) in our LME analyses (Table 4).
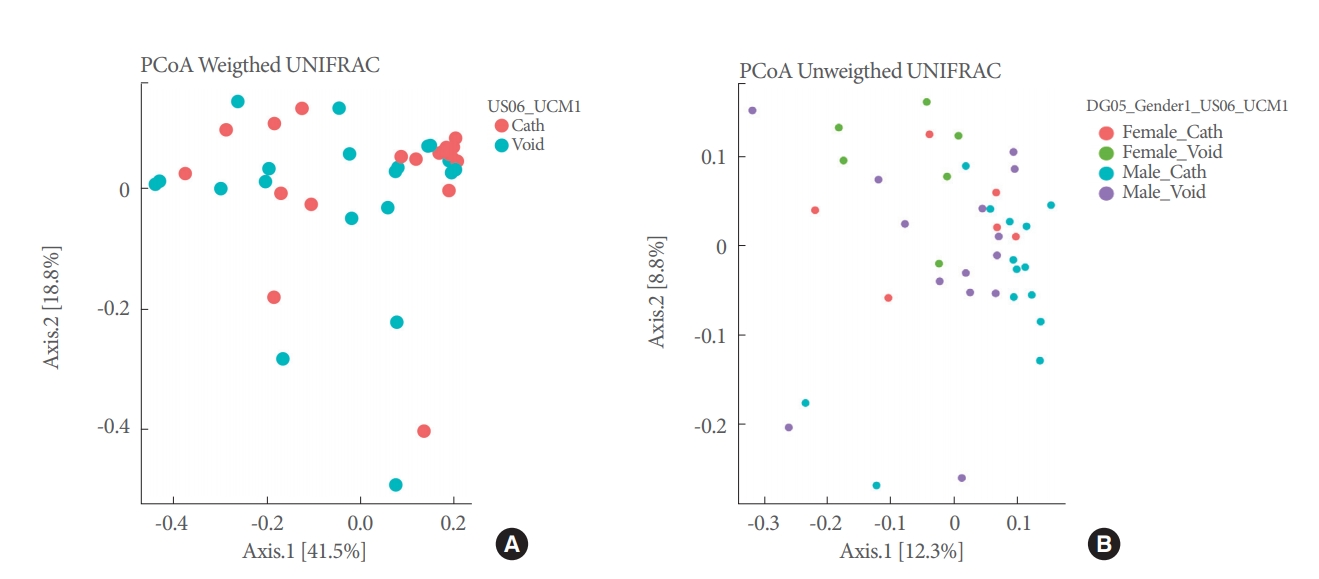
PCoAs of Unifrac (unweighted and weighted) distances showed partial segregation of the microbial communities from each group (Fig. 3), but the adonis analyses only detected significant differences in beta-diversity (intersample) between urethra and bladder samples for the Unifrac unweighted distance (P=0.043).
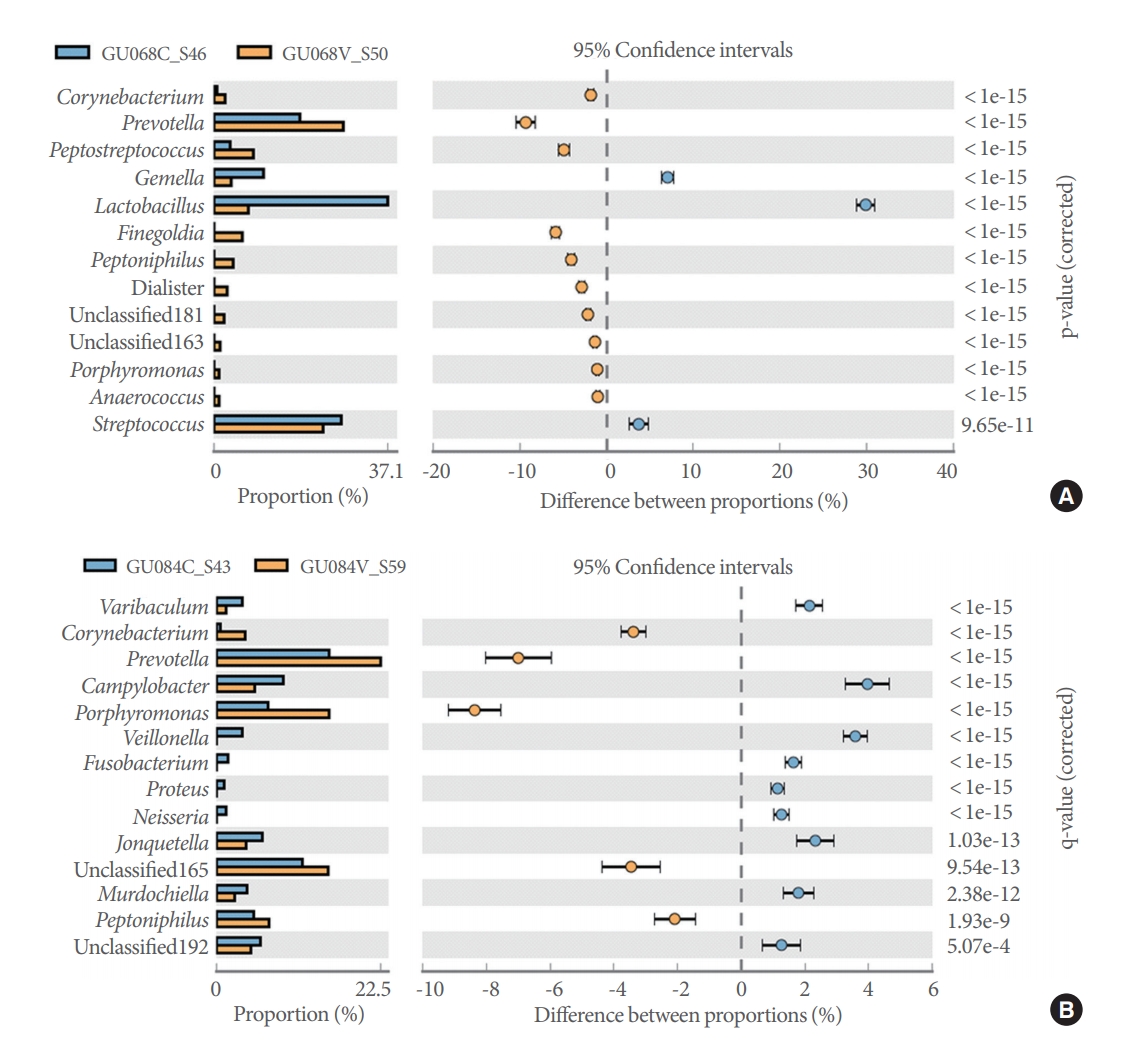
Pairwise comparisons of catheterized and voided samples from each patient using the Fisher exact test detected 6 to 20 genera showing significant differences in abundance across the 20 patients. Representative diversity plots for a 20-year-old male and 33-year-old female are demonstrated (Fig. 4A, B).
Male Versus Female Samples
Microbial mean proportions of Lactobacillus, Veillonella, Staphylococcus, Enterobacter sp, and Haemophilus varied greatly (fold change >2) between sexes (Table 4). Male samples showed the highest abundance of Streptococcus, Veillonella, Staphylococcus, Gardnerella, Enterobacter sp, Neisseria, and Haemophilus, while female samples showed the highest abundance of Lactobacillus and Prevotella. Our LME analyses only showed significant differences in abundance for Lactobacillus (P=0.021).
Alpha-diversity (intrasample) indices (Shannon, ACE, and PD) did not vary significantly across sex phenotypes (Fig. 2, Table 4). PCoAs of Unifrac (unweighted and weighted) distances also showed partial segregation of the microbiotas from each group (Fig. 3), while the adonis analyses showed significant differences in beta-diversity (intersample) between males and females for both Unifrac distances (P≤0.031). Our LME and adonis analyses did not show significant results for any of the urine collection method or sex interactions assessed. Moreover, our sex-based analyses must be interpreted with caution given the unequal sample size of females (12 samples) and males (28 samples) in our study.
DISCUSSION
In this pilot study, we analyzed urine microbiome 16S rRNA sequences from asymptomatic individuals with no known genitourinary pathology to investigate the urine microbiome in asymptomatic females and males without bladder pathology. We sought to determine whether the urine microbiome differs by urine collection method (voided versus catheterized). This goal was based on a hypothesis that sampling method would allow discrimination between the microbiome of the urethra and the bladder and between sexes. Within our cohort of asymptomatic young adult volunteers, we identified Firmicutes as the most commonly represented phylum, with Lactobacillus being the predominate genus in females and Streptococcus the predominate genus in males. Only the former varied significantly (P=0.021) across sexes, but significant (P≤0.031) differences in microbiome composition and structure (Fig. 3) were found between males and females (Table 4). This work confirms our earlier pilot data in which we demonstrated a sex-specific urine microbiome predominated by Lactobacillus and Streptococcus at the genus level, in females and males, respectively [21]. Comparisons between samples obtained by catheterization and by voiding suggest that the healthy microbiome of the urethra significantly (P=0.043) differs from that of the bladder in alpha-diversity (Fig. 2), composition (Fig. 3), and the abundances of several genera (Table 4).
Lewis et al. [22] analyzed the human urinary microbiome in voided midstream urine samples from 6 males and from 10 females (ages 26–90 years; median, 64.5 years) without symptoms, noting a “core” urinary microbiome that differed by sex and fluctuated with age. They saw a trend toward more bacterial heterogeneity among females (median number of genera, 21) than males (median number of genera, 11). Three-quarters of the samples in both sexes contained >50% abundance of Firmicutes, while only female samples had members of Acinetobacter and Bacteroides which were absent among males [22]. Although the number of genera did not vary by age, Jonquetella, Parvimonas, Proteiniphilum, and Saccharofermentans were only seen in those 70 years old and greater, irrespective of sex [22]. Bacterial diversity was highly variable between individuals regardless of age and sex; however, the data—particularly from females—suggest a “core” microbiome of 23 genera which fluctuates in total amount of bacteria as well as the number and type of “cohabitating” genera. While Lewis et al. [22] studied healthy adults from a broad range of ages, our cohort—with a median age of 33 years—is representative of the young-to-middle aged adult population, most with negative urine culture by standard cultivation techniques. While noting the likely contribution of urethral and/or perineal contaminants to their urine samples, the authors asserted their methods to be pragmatic given the asymptomatic nature of their subjects. However, they proposed that future studies might include an assessment of urethral microbiota to account for these regional differences in bacterial communities. Ideally, comparisons of suprapubic aspirated urine to urethral sampling could help better define differences in these microbiomes. However, ethical approval of suprapubic aspiration in healthy persons is difficult in the United States.
It is widely known that culture-independent sequencing methods identify microbes that are not routinely detectable by culture. 16S rRNA single molecule, real-time sequencing yielded more genera than would have been potentially identified by standard culture techniques. Although the clinical utility of identifying such cryptic microbes is uncertain, it is hypothe-sized that these microbes might participate in UTI recurrence/ persistence, specific infection-associated symptoms, and/or antimicrobial resistance. By extension, knowledge of what constitutes a “healthy” urine microbiome might afford opportunity to manipulate a dysbiotic community in symptomatic individuals. Indeed, while NGS of 16S amplicons identified microbiota in each individual studied, only 4 participants demonstrated bacteria by standard cultivation techniques. In each case, the number of colonies did not meet established clinical thresholds to make the diagnosis of UTI, particularly since none had urinary complaints, thus suggesting these were contaminated specimens. However, 16S rRNA sequencing identified the presence of these microbes in every case as well as others not identified through culture, which reflects the robust sensitivity of sequencing.
Sex differences in the core microbiome clearly exist. We have convincingly shown twice before [21,23], and in this work, that the genus Lactobacillus predominates in the microbiome of healthy women. Among men, our findings appear contradictory with earlier work identifying predominately Corynebacterium, while our most recent studies have identified Streptococcus as the predominate genus. In Fouts et al. [23], Corynebacterium seemed to be a hallmark of the healthy male urine microbiome since its prevalence was diminished among asymptomatic males with increasing severity of neuropathic bladder dysfunction. Dong et al. [24] similarly identified Corynebacterium in urethral swab and clean-voided samples of healthy males, suggesting that Corynebacterium resides in the urethra and bladder, and that it is a constituent of the healthy male urine microbiome.
Discrepancy in the predominate urine microbe(s) found in males can be explained by differences in NGS platform, 16S rRNA regions sequenced, analytical pipelines and 16S reference databases. In Fouts et al. [23], we investigated voided urine samples from healthy asymptomatic adults by coupling 454 pyrosequencing of the hypervariable V1-V3 regions of the 16s rRNA gene with the mothur pipeline and the Silva database (2007 release), which was an established methodology at the time for genus level characterization of the microbiome. Our follow-up work, we re-analyzed the samples described in Fouts et al. [23], using PathoScope analysis of LTP115 and Silva (119 Ref NR 99) databases. In that study, we confirmed that the male healthy voided urine microbiome was predominately comprised of Streptococcus at the genus level and Staphylococcus haematolyticus as the predominate sp. In the current study, a new cohort of asymptomatic controls has been investigated using MiSeq sequences of the hypervariable V4 region of the 16s rRNA gene, the mothur pipeline, as in our first study, and the Silva database (2015 release), and confirmed that Streptococcus, and not Corynebacterium, is the predominate genus in males.
Our previous work [21] and the findings of this study, lend credence to the hypothesis that, especially in females, lactobacilli are important constituents of the healthy urine microbiome. Such a microbiome may very well be characterized by Lactobacillus crispatus during health and Lactobacillus iners during disease as has been seen in the vagina [25]. We further hypothesize that the normal urinary microbiomes in both females and males, are not only constitutively different between sexes, but also differ regionally within the urinary tract as a barrier to invasion by pathogenic organisms. It is also possible that regional differences in the urinary tract microbiome reflect occupation of specific ecological niches associated with the tissue microenvironments of the urethra vs. bladder. This is a plausible hypothesis given that the urethra is less exposed to urine than the bladder, and the urethra features a continuum of squamous and urothelial epithelium, whereas the bladder is entirely lined with urothelium. Differing mechanical forces in the urethra versus bladder may also select for bacteria with specific adhesive and tissue-invasive properties.
In conclusion, the impact of urine collection method on microbiome studies is relevant to future research as genetic analyses add to our understanding of urinary health and disease. Dong et al. [24] demonstrated that the microbiota of samples obtained by urethral swab and voided urine in males presenting to a sexually transmitted infection (STI) clinic yielded similar proportions of classifiable taxa irrespective of whether the individuals did (n=10) or did not (n=22) have STI. However, it is entirely likely that voided samples represent a mixture of bacteria from the urethra and bladder, and less likely that the regional microbiota of the urethra and bladder are similar.